问题描述:荧光定量[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]扩增效率低的原因?解答:[align=left][font=宋体][color=black]可能的原因有:[/color][/font][/align][align=left][font=宋体][color=#262626][back=white]([/back][/color][/font][font='Times New Roman','serif'][color=#262626][back=white]1[/back][/color][/font][font=宋体][color=#262626][back=white])反应试剂中部分成分特别是荧光染料降解。[/back][/color][/font][/align][align=left][font=宋体][color=#262626][back=white]([/back][/color][/font][font='Times New Roman','serif'][color=#262626][back=white]2[/back][/color][/font][font=宋体][color=#262626][back=white])反应条件不够优化,可适当降低退火温度或改为三步扩增法。[/back][/color][/font][/align][align=left][font=宋体][color=#262626][back=white]([/back][/color][/font][font='Times New Roman','serif'][color=#262626][back=white]3[/back][/color][/font][font=宋体][color=#262626][back=white])反应体系中有[/back][/color][/font][font='Times New Roman','serif'][color=#262626][back=white][url=https://insevent.instrument.com.cn/t/jp]PCR[/url][/back][/color][/font][font=宋体][color=#262626][back=white]反应抑制物,一般是加入模板时所引入,应先把模板适度稀释,再加入反应体系中,以减少抑制物的影响。[/back][/color][/font][/align]以上内容来自仪器信息网《PCR实战宝典》
[font=宋体][size=14.0pt]荧光定量[/size][/font][size=14.0pt]PCR[/size][font=宋体][size=14.0pt]实验失败原因分析 ——荧光信号检测出了问题[/size][/font][font=宋体][size=14.0pt]近日某实验室来电,反映其荧光定量[/size][/font][size=14.0pt]PCR[/size][font=宋体][size=14.0pt]仪在成功检测了一次病毒核酸后,连续几次检测均失败,表现为阳性对照也无[/size][/font][size=14.0pt]Ct[/size][font=宋体][size=14.0pt]值,请求给予技术支持。[/size][/font][size=14.0pt]1[/size][font=宋体][size=14.0pt]背景[/size][/font][font=宋体][size=14.0pt]该实验室荧光定量[/size][/font][size=14.0pt]PCR[/size][font=宋体][size=14.0pt]仪系进口品牌,今年[/size][/font][size=14.0pt]4[/size][font=宋体][size=14.0pt]月份安装调试,当时检测结果较理想。[/size][/font][size=14.0pt]5[/size][font=宋体][size=14.0pt]月份该实验室进行了一次病毒核酸检测,提取采用柱式[/size][/font][size=14.0pt]DNA[/size][font=宋体][size=14.0pt]提取试剂盒,未设阴性对照和阳性对照;扩增采用配套的核酸扩增试剂,设置了阴性对照和阳性对照,检测结果较理想。但在后续的检测工作中,却连续多次实验失败,表现为整个扩增过程荧光信号均为一条几乎无起伏的直线。[/size][/font][size=14.0pt]2[/size][font=宋体][size=14.0pt]思路[/size][/font][font=宋体][size=14.0pt]影响荧光定量[/size][/font][size=14.0pt]PCR[/size][font=宋体][size=14.0pt]检测结果的因素可分为三个阶段,一是核酸提取,二是核酸扩增,三是荧光信号检测。为了分析是哪个阶段出了问题,我们设计了以下方案:[/size][/font][font=宋体][size=14.0pt]第一步:利用已知结果的核酸[/size][/font][size=14.0pt]+[/size][font=宋体][size=14.0pt]该实验室的扩增试剂,看有无检测结果。如有则说明核酸提取环节存在问题,如无则说明可能是核酸扩增环节或荧光信号检测存在问题,继续进行第二步分析;[/size][/font][font=宋体][size=14.0pt]第二步:利用已知结果的核酸[/size][/font][size=14.0pt]+[/size][font=宋体][size=14.0pt]经验证可以得到结果的扩增试剂(本实验室在用的扩增试剂及引物探针),看有无检测结果。如有则说明核酸提取环节存在问题,如无则说明可能是荧光信号检测存在问题。[/size][/font][size=14.0pt]3[/size][font=宋体][size=14.0pt]分析验证[/size][/font][font=宋体][size=14.0pt]方案确定后,带上本实验室已知结果的核酸(包括[/size][/font][size=14.0pt]Ct[/size][font=宋体][size=14.0pt]值[/size][/font][size=14.0pt]25[/size][font=宋体][size=14.0pt]、[/size][/font][size=14.0pt]28[/size][font=宋体][size=14.0pt]、[/size][/font][size=14.0pt]31[/size][font=宋体][size=14.0pt]、[/size][/font][size=14.0pt]34[/size][font=宋体][size=14.0pt]及阴性),以及本实验室在用的扩增试剂及引物探针,到该实验室开展分析验证。[/size][/font][font=宋体][size=14.0pt]第一步:利用已知结果的核酸[/size][/font][size=14.0pt]+[/size][font=宋体][size=14.0pt]该实验室的扩增试剂上机检测,结果荧光信号仍为一条几乎无起伏的直线。说明可能是核酸扩增环节或荧光信号检测存在问题。为明确究竟是何原因造成实验失败,继续开展第二步分析验证。[/size][/font][font=宋体][size=14.0pt]第二步:利用已知结果的核酸[/size][/font][size=14.0pt]+[/size][font=宋体][size=14.0pt]经验证可以得到结果的扩增试剂上机检测,结果荧光信号还为一条几乎无起伏的直线。因本次使用的扩增试剂和引物探针是本实验室一直在用的,已知可以得到正确的扩增结果,所以初步判断可能是荧光信号检测存在问题。[/size][/font][font=宋体][size=14.0pt]到此,分析验证告一段落,实验失败极有可能是荧光定量[/size][/font][size=14.0pt]PCR[/size][font=宋体][size=14.0pt]仪出现故障,最大可能是荧光信号检测存在问题。目前,该实验室已电告设备厂家,要求派工程师上门维修。[/size][/font][size=14.0pt]4[/size][font=宋体][size=14.0pt]思考[/size][/font][font=宋体][size=14.0pt]荧光定量[/size][/font][size=14.0pt]PCR[/size][font=宋体][size=14.0pt]实验失败看似非常头痛,实则不然。只要理清思路,逐一分析验证核酸提取、核酸扩增、荧光信号检测各个环节,即可快速判断问题所在。[/size][/font][font=宋体][size=14.0pt]另外还有一点不吐不快的感受,作为一个世界著名品牌产品,实验室选择它的出发点就是该设备的准确性和稳定性,但这台设备只进行过一次检测就出现故障,实属不该。[/size][/font][font=宋体][size=14.0pt][font=&]5[/font]续[/size][/font][font=宋体][size=14.0pt]最新消息,据该实验室反馈,厂方工程师上门检修,发现是作为荧光光源的灯泡坏了。更换灯泡后,该实验室已能检测出正确结果。[/size][/font]

【参数解读】基因扩增仪(PCR)的技术参数解读与使用PCR技术的基本原理PCR技术的基本原理 类似于DNA的天然复制过程,其特异性依赖于与靶序列两端互补的寡核苷酸引物。PCR由变性--退火--延伸三个基本反应步骤构成:①模板DNA的变性:模板DNA经加热至93℃左右一定时间后,使模板DNA双链或经PCR扩增形成的双链DNA解离,使之成为单链,以便它与引物结合,为下轮反应作准备;②模板DNA与引物的退火(复性):模板DNA经加热变性成单链后,温度降至55℃左右,引物与模板DNA单链的互补序列配对结合;③引物的延伸:DNA模板--引物结合物在TaqDNA聚合酶的作用下,以dNTP为反应原料,靶序列为模板,按碱基配对与半保留复制原理,合成一条新的与模板DNA 链互补的半保留复制链重复循环变性--退火--延伸三过程,就可获得更多的“半保留复制链”,而且这种新链又可成为下次循环的模板。每完成一个循环需2~4分钟, 2~3小时就能将待扩目的基因扩增放大几百万倍。请您来解析:1、普通PCR仪,梯度PCR仪,原位PCR仪,实时荧光定量PCR仪四类仪器有何异同?http://ng1.17img.cn/bbsfiles/images/2015/02/201502251536_536341_1608710_3.png2、我们应该如何根据检测需求来选购PCR的四类仪器?根据检测的定量要求来选择仪器类型。3、PCR的核心参数都有哪些?核心参数是温度控制。4、梯度PCR仪可以分离不同DNA的片段吗?与普通PCR的异同在哪?梯度PCR可以分离不同DNA片段,与普通PCR仪不同的地方在于。可以设置一系列的梯度退火温度进行扩增,从而一次性PCR扩增就可以筛选出表达量高的最适合退火温度进行有效的扩增。主要用于研究未知DNA退火温度的扩增,这样可节省试验时间、提高实验效率,又节约实验成本。5、使用过程中常见的问题PCR反应的最大特点是具有较大扩增能力与极高的灵敏性,但令人头痛的问题是易污 染,极其微量的污染即可造成假阳性的产生. 一、污染原因 (一)标本间交叉污染:标本污染主要有收集标本的容器被污染,或标本放置时,由于 密封不严溢于容器外,或容器外粘有标本而造成相互间交叉污染;标本核酸模板在提 取过程中,由于吸样枪污染导致标本间污染;有些微生物标本尤其是病毒可随气溶胶 或形成气溶胶而扩散,导致彼此间的污染. (二)PCR试剂的污染:主要是由于在PCR试剂配制过程中,由于加样枪、容器、双蒸水 及其它溶液被PCR核酸模板污染. (三)PCR扩增产物污染.这是PCR反应中最主要最常见的污染问题.因为PCR产物拷贝量 大(一般为1013拷贝/ml),远远高于PCR检测数个拷贝的极限,所以极微量的PCR产物 污染,就可造成假阳就可形成假阳性. 还有一种容易忽视,最可能造成PCR产物污染的形式是气溶胶污染;在空气与液体面摩 擦时就可形成气溶胶,在操作时比较剧烈地摇动反应管,开盖时、吸样时及污染进样 枪的反复吸样都可形成气溶胶而污染.据计算一个气溶胶颗粒可含48000拷贝,因而由 其造成的污染是一个值得特别重视的问题. (四)实验室中克隆质粒的污染:在分子生物学实验室及某些用克隆质粒做阳性对照的 检验室,这个问题也比较常见.因为克隆质粒在单位容积内含量相当高,另外在纯化 过程中需用较多的用具及试剂,而且在活细胞内的质粒,由于活细胞的生长繁殖的简 便性及具有很强的生命力.其污染可能性也很大. 二、污染的监测 一个好的实验室,要时刻注意污染的监测,考虑有无污染是什么原因造成的污染,以 便采取措施,防止和消除污染. 对照试验 1.阳性对照:在建立PCR反应实验室及一般的检验单位都应设有PCR阳性对照,它是PCR 反应是否成功、产物条带位置及大小是否合乎理论要求的一个重要的参考标志.阳性 对照要选择扩增度中等、重复性好,经各种鉴定是该产物的标本,如以重组质粒为阳 性对照,其含量宜低不宜高(100个拷贝以下).但阳性对照尤其是重组质粒及高浓度阳 性标本,其对检测或扩增样品污染的可能性很大.因而当某一PCR试剂经自己使用稳 定,检验人员心中有数时,在以后的实验中可免设阳性对照. 2.阴性对照:每次PCR实验务必做阴性对照.它包括①标本对照:被检的标本是血清就用 鉴定后的正常血清作对照;被检的标本是组织细胞就用相应的组织细胞作对照.②试剂 对照:在PCR试剂中不加模板DNA或RNA,进行PCR扩增,以监测试剂是否污染. 3.重复性试验 4.选择不同区域的引物进行PCR扩增 三、防止污染的方法 (一)合理分隔实验室:将样品的处理、配制PCR反应液、PCR循环扩增及PCR产物的鉴定 等步骤分区或分室进行,特别注意样本处理及PCR产物的鉴定应与其它步骤严格分开. 最好能划分①标本处理区;②PCR反应液制备区;③PCR循环扩增区;④PCR产物鉴定区. 其实验用品及吸样枪应专用.实验前应将实验室用紫外线消毒以破坏残留的DNA或RNA. (二)吸样枪:吸样枪污染是一个值得注意的问题.由于操作时不慎将样品或模板核酸吸 入枪内或粘上枪头是一个严重的污染源,因而加样或吸取模板核酸时要十分小心,吸 样要慢,吸样时尽量一次性完成,忌多次抽吸,以免交叉污染或产生气溶胶污染. (三)预混和分装PCR试剂:所有的PCR试剂都应小量分装,如有可能,PCR反应液应预先 配制好,然后小量分装,-20℃保存.以减少重复加样次数,避免污染机会.另外,PCR试剂,PCR反应液应与样品及PCR产物分开保存,不应放于同一冰盒或同一 冰箱. (四)防止操作人员污染,使用一次性手套、吸头、小离心管应一次性使用. (五)设立适当的阳性对照和阴性对照,阳性对照以能出现扩增条带的最低量的标准病 原体核酸为宜,并注意交叉污染的可能性,每次反应都应有一管不加模板的试剂对照 及相应不含有被扩增核酸的样品作阴性对照. (六)减少PCR循环次数,只要PCR产物达到检测水平就适可而止. (七)选择质量好的Eppendorf管,以避免样本外溢及外来核酸的进入,打开离心管前 应先离心,将管壁及管盖上的液体甩至管底部.开管动作要轻,以防管内液体溅出. 常见问题分析与对策 PCR产物的电泳检测时间 一般为48h以内,有些最好于当日电泳检测,大于48h后带型不规则甚致消失。 假阴性,不出现扩增条带 PCR反应的关键环节有①模板核酸的制备,②引物的质量与特异性,③酶的质量及, ④PCR循环条件。寻找原因亦应针对上述环节进行分析研究。 模板:①模板中含有杂蛋白质,②模板中含有Taq酶抑制剂,③模板中蛋白质没有消 化除净,特别是染色体中的组蛋白,④在提取制备模板时丢失过多,或吸入酚。⑤模 板核酸变性不彻底。在酶和引物质量好时,不出现扩增带,极有可能是标本的消化处 理,模板核酸提取过程出了毛病,因而要配制有效而稳定的消化处理液,其程序亦应 固定不宜随意更改。 酶失活:需更换新酶,或新旧两种酶同时使用,以分析是否因酶的活性丧失或不够而 导致假阴性。需注意的是有时忘加Taq酶或溴乙锭。 引物:引物质量、引物的浓度、两条引物的浓度是否对称,是PCR失败或扩增条带不 理想、容易弥散的常见原因。有些批号的引物合成质量有问题,两条引物一条浓度 高,一条浓度低,造成低效率的不对称扩增,对策为:①选定一个好的引物合成单 位。②引物的浓度不仅要看OD值,更要注重引物原液做琼脂糖凝胶电泳,一定要有 引物条带出现,而且两引物带的亮度应大体一致,如一条引物有条带,一条引物无条 带,此时做PCR有可能失败,应和引物合成单位协商解决。如一条引物亮度高,一条 亮度低,在稀释引物时要平衡其浓度。③引物应高浓度小量分装保存,防止多次冻融 或长期放冰箱冷藏部分,导致引物变质降解失效。④引物设计不合理,如引物长度不 够,引物之间形成二聚体等。 Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低PCR扩增的特 异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。 反应体积的改变:通常进行PCR扩增采用的体积为20ul、30ul、50ul。或100ul,应用多 大体积进行PCR扩增,是根据科研和临床检测不同目的而设定,在做小体积如20ul 后,再做大体积时,一定要模索条件,否则容易失败。 物理原因:变性对PCR扩增来说相当重要,如变性温度低,变性时间短,极有可能出 现假阴性;退火温度过低,可致
俺公司新建一实验室,做酶免,胶金,核酸扩增,请问需要哪些仪器

如下手持式基因与核酸扩增分析仪的市场需求怎么样?[img=,690,975]https://ng1.17img.cn/bbsfiles/images/2019/07/201907121534081873_2288_2963345_3.jpg!w690x975.jpg[/img]
1971年Kleppe等人在Journal of molecular biology上发表文章首次准确、精炼、客观的阐述了PCR方法,1976年一种从嗜热水生菌(Thermus aquaticus)分离得到的热稳定的DNA依赖的DNA聚合酶的应用大大增加了PCR的效率。而现今所发展出来的PCR则是源于由Saiki和Mullis等人于1988年发表在Science上的一篇论文,Mullis当时服务于Perkin Elmer(PE)公司,因此PE公司在PCR界有着特殊的地位。后来PE被Applied Biosystems Inc.(ABI)公司收购、分拆、再转卖,而PCR的专利和倍受信赖的PCR仪器生产和销售就留在ABI名下。到如今,PCR方法愈发趋向自动化,并从中衍生出更多的新技术方法,可以说,PCR技术是支撑现代分子生物学发展的一块重要基石。这种技术的广泛应用催生了一个庞大的市场,多个公司均有各种类型的商品化PCR仪出售。PCR的专利目前依然掌握在ABI和Roche(罗氏)两大公司手中,去年业界颇为引人瞩目ABI诉MJ公司侵犯侵犯PCR仪知识产权案最终以MJ败诉并宣布破产、最终被Bio-rad收购暂告一段落。其后还会不会有后继的故事还需拭目以待。 PCR原理 DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解链成单链,在DNA聚合酶的作用下,以单链为模版,根据碱基互补配对原则复制成新的单链,与模版配对成为双链分子挎贝。在体外实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,并设计与模板DNA的5’端结合的两条引物,加入DNA聚合酶、dNTP就可以完成特定基因的体外复制,多次重复“变性解链—退火—合成延伸”的循环就可以以几何级数大量扩增特定的基因。 发现耐热DNA聚合酶对于PCR的应用有里程碑的意义,该类酶可以耐受90℃以上的高温而不失活,不需要每个循环加酶,使PCR技术变得非常简捷、同时也大大降低了成本,PCR技术得以大量应用,并逐步应用于临床。 从PCR原理可以看出,PCR仪的关键是升降温的步骤。现在偶尔还能听到一些前辈们笑谈早年的PCR实验如何在3个水浴锅中完成的趣闻。经过不断改进,今天的PCR已经越来越完善和智能化。出于市场推广的战略需要,各厂家的PCR仪型号不同,着力宣传的技术指标和参数也不尽统一,编者在这里简单列出选购时我们认为应该考虑的常用指标,希望有助于大家选购PCR仪的选购技巧。PCR仪介绍及其选购 PCR仪的种类总体来说可以分为两大类:PCR扩增仪和实时荧光定量PCR仪,普通的PCR扩增仪又衍生出带梯度PCR功能的梯度PCR仪、和带原位扩增功能的原位PCR仪等等。1996年由ABI公司首先推出将扩增和检测融为一体的实时荧光定量PCR仪,此后很多公司如Eppendorf、ROCHE、Corbett、MJ等等都先后推出不同款式的定量PCR仪。
动物疫病荧光定量PCR检测方法的模块化构建及实践1 背景近几十年来,动物疫病的发生与发展越来越趋向频繁、复杂,造成的危害也不断加重。究其原因:一是动物数量大幅度增加,集约化程度不断提高。据《中国统计年鉴》:1990年底全国猪、羊存栏分别为5668万头、11362万头,2015年底则分别为45803万头、31174万头,分别增加7.08倍、1.74倍;二是动物调运频繁、距离不断延长。以前动物调运仅限省内或周边省市,现在动辄上千公里甚至数千公里。据报道:“从2013年底到2014年的上半年,我国新疆、甘肃、内蒙古、宁夏等省区接连爆发了较为严重的小反刍兽疫疫情,而后疫情逐渐扩展地全国20余个省份,造成了严重的经济、社会影响”(《中国畜牧业》2014年第21期《小反刍兽疫爆发对我国肉羊产业发展的影响》)。三是人畜共患病种类不断增加。2012年6月加拿大Haroling教授报告,目前人类已发现1000多种动物病原体,其中有600种存在于家畜之中,在这600种病原体中有40%的病原体为人畜共患病。从国内情况看,进入21世纪以来,禽流感(H5亚型、H7N9)、甲型H1N1流感、SARS、猪链球菌病2型、布氏杆菌病、结核、乙脑、狂犬病等人畜共患病不断发生。为确保公共卫生安全,以及畜牧业健康发展,有必要实施动物疫病的核酸监测,尽早发现疫情隐患并予以及时处置,避免重大动物疫情及公共卫生安全事件发生。2 常用试剂盒的缺点目前,动物疫病(主要指病毒病)核酸检测一般采用商品化荧光PCR试剂盒,多由双组分组成且独立保存,使用前按一定比例(一般PCR反应液与酶混合液体积比为9:1)混匀后用于检测。这种试剂盒存在几个缺点:一是只能用于某一特定的动物疫病检测,如果检测任务中途发生大幅度削减,则试剂盒只能作报废处理;二是试剂盒中有的组分容易缺斤少两,尤其是酶混合液更是如此。每个样品约需酶混合液1.2μL,一般10T试剂盒装量为15μL,50T试剂盒酶混合液装量为70[font=宋体-方正超大字符集][size=18px]μL[/size][/font],按理说应该足够使用。但实际检测中会出现10T试剂盒酶混合液只能检测7T,50T试剂盒酶混合液只能检测40T,严重影响检测工作的正常开展。分析其原因,可能与容器密闭不严、酶混合液中的水分在冰冻状态升华等有关;三是试剂盒中的引物探针信息缺失。出于保护商业秘密考虑,几乎所有试剂盒均对引物探针的具体序列进行保密。作为第三方检测机构,在开展检测工作时,应优先使用标准方法,并确保使用标准的有效版本。在使用标准方法前,应进行证实。在使用非标准方法(含自制方法)前,应进行确认。但由于试剂盒厂家不提供引物探针序列,检测机构无法判定使用试剂盒开展的检测是否符合标准方法的要求。另外,商品化试剂盒一般免费附带核酸提取试剂,但提取试剂根据检测项目的不同,一般为DNA专用提取试剂或RNA专用提取试剂。当一批样品要检测多种DNA及RNA病毒项目时,需要同时进行两次核酸提取,不仅费时费力且浪费试剂。由于上述缺点的存在,给动物疫病检测带来了不少麻烦。3 模块化检测方法的构建思路针对商品化试剂盒存在的缺点,进行了模块化检测方法的构建。总体思路是:将动物疫病荧光定量PCR检测分为三个模块,核酸提取采用DNA/RNA共提试剂,核酸扩增采用预混液+引物探针方式,质控品利用抗原或商品化疫苗自制。具体思路如下:——核酸提取采用DNA/RNA共提试剂;——引物探针严格按照标准方法,委托生物工程公司合成;——反应体系中其他成分,采购商品化qPCR预混液或RT-qPCR预混液(含逆转录酶);——质控品:阳性对照采用病毒抗原、商品化疫苗等,测定结果后按一定比例稀释,保存备用。——反应体系:一律固定为20μL,具体如下:[table][tr][td]DNA扩增[/td][td]RNA扩增[/td][/tr][tr][td=1,2]qPCR预混液(2×) 10μL[/td][td]RT-qPCR预混液(2×) 10μL[/td][/tr][tr][td]逆转录酶 1μL[/td][/tr][tr][td]ROX 0.4μL[/td][td]ROX 0.4μL[/td][/tr][tr][td]引物探针 适量[/td][td]引物探针 适量[/td][/tr][tr][td]水 补足18μL[/td][td]水 补足18μL[/td][/tr][tr][td]DNA模板 2μL[/td][td]RNA模板 2μL[/td][/tr][/table]——反应条件首先按照标准中的条件开展摸索,如达到预期效果则予以固定;如效果不佳则参照预混液及引物探针推荐条件进行适当调整。根据上述思路,一个实验室只需配备DNA/RNA共提试剂、qPCR预混液、RT-qPCR预混液共3种试剂,加上特定的引物探针,即可开展多种动物疫病的检测。此配置有以下优点:一是试剂通用性强。实验室只需配备DNA/RNA共提试剂、qPCR预混液(针对DNA病毒)、RT-qPCR预混液(针对RNA病毒)三种主要试剂,以及不同的引物探针,即可开展几种甚至几十种检测项目;二是灵活方便。根据动物疫病的发生发展,实验室可以随时调整检测项目和检测数量。以RT-qPCR预混液为例,不仅可以用于检测禽流感,也可以用于检测口蹄疫、猪瘟、蓝耳病等项目。实验室常备一定数量的预混液,当某个项目检测量突然增加时,可利用预混液的通用特性,迅速完成检测任务,尔后再采购预混液进行补充。当某个项目因故取消时,也可将预混液用于其他项目的检测,不致于造成大量浪费。三是方便开展项目检测。当突发新的重大疫情,上级有关部门公布引物探针序列及扩增程序后,可以委托生物工程公司紧急合成引物探针,利用现有的预混液,在几天内即可具备新方法的检测能力,而这一点是成品试剂盒很难做到的。4 质控品制备对于病毒核酸荧光定量PCR检测,每次实验都必须设立阴性对照和阳性对照。一般情况下,在核酸提取和核酸扩增,需分别设立阴性对照和阴性对照,也就是说每次检测至少需要设立E-(核酸提取阴性对照)、E+(核酸提取阳性对照)、R-(核酸扩增阴性对照)、R+(核酸扩增阳性对照)共4个质控。在开展多次检测,结果稳定后,可以仅使用E-/E+或R-/R+。阴性对照可用DEPC水,阳性对照是否可以利用商品试剂盒附带的质控品?经过部分项目的检测,证明大部分商品试剂盒附带的质控品,均无法作为实验室自制的准标准方法的阳性对照。原因可能是商品试剂盒质控品系根据其专有的引物探针(与检测标准不一致)扩增片段制成质粒,只能使用其特有的引物探针才能检测到。因此,需要自制阳性对照用于质控。对于基层实验室来说,制备质粒或者假病毒非常困难,较可靠的办法是直接利用全病毒来制备阳性对照,具体思路如下:——对于禽流感、新城疫、口蹄疫等项目,由于商品化试剂盒附带有全病毒抗原,可以尝试使用这些抗原来制备阳性对照;——对于蓝耳病、伪狂犬、圆环病毒、小反刍兽疫等项目,由于已经有商品化疫苗上市,可以尝试使用商品化疫苗来制备;——对于部分既无全病毒抗原又无商品化疫苗的检测项目,在不违反法律法规及部门规定的情况下,可以利用已确认阳性的病毒核酸,适当稀释后作为核酸扩增阳性对照(R+);或者将病毒核酸用已灭活的阴性血浆作适当稀释,复原成模拟阳性样品,作为核酸提取阳性对照(E+)。5 应用根据上述思路,进行了相关实验,取得了初步成果。现简要汇报如下:——核酸提取采用FastPure Viral DNA/RNA Mini Kit试剂盒,南京诺维赞生产,可以实现多种样品的DNA/RNA共提;——引物探针按照相应国家标准提供的序列,委托上海生工合成,引物稀释成10μmol/L、探针稀释成5μmol/L,分装成小包装冷冻保存备用;——qPCR预混液采用AceQ Universal U+ Probe Master Mix V2,RT-qPCR预混液采用2× One Step Q Probe Mix,逆转录酶采用One Step Q Probe Enzyme Mix,均为南京诺维赞生产;——质控品:禽流感选用血凝抑制抗原,口蹄疫选用液相阻断ELISA抗原,其他选用商品化疫苗,测定结果后用灭菌水稀释至Ct值约25~30,冷冻保存备用;——反应体系:设置为20μL,预混液(2×)10μL、逆转录酶1μL、ROX 0.4μL为固定量,引物探针加入量根据检测标准中的终浓度予以换算,加灭菌水补足18μL,DNA或RNA模板为2μL;——反应条件:绝大多数项目直接套用检测标准中的反应条件,效果良好不需要调整。6 讨论6.1 检测工作更加简捷高效模块化检测方法,极大地方便了检测工作的开展。目前实验室经常开展的检测项目有10余个,如果采用商品化试剂盒,则每个检测项目需专用的试剂盒,采购、保存和使用等各个环节,均显得很不方便。改用模块化检测方法后,只需要1种核酸提取试剂和2种扩增试剂,加上各检测项目专用的引物探针,即可开展多个项目的检测。6.2 适用性有待进一步验证尽管在构建新方法时,对适用性进行了尽可能多的验证,如对于动物A型流感,就验证了H5亚型Re-4、Re-5、Re-6、Re-7、Re-8、Re-10、Re-11、Re-12,以及H7亚型、H7N9、H7N9Re-1、H7N9Re-2等毒株的抗原,均证实有效。但对于其他毒株,尤其是较新型的变异株是否有效,尚有待进一步观察和验证。6.3 关于质控品质控品在某个检测方法中,起到非常重要的作用。E-出现阳性结果可能是核酸提取过程中受到污染,E+出现阴性结果可能是核酸提取试剂失效或提取过程出现问题,R-出现阳性结果可能是核酸扩增试剂受到污染,R+出现阴性结果可能是核酸扩增试剂失效。根据E-/E+/R-/R+的扩增结果,大致可以判断核酸提取和核酸扩增过程是否出现问题。阴性对照一般可用灭菌水代替,阳性对照可选用扩增效果较好的抗原或疫苗。6.4 规范操作避免污染由于阳性对照使用的是全病毒抗原或疫苗,一旦造成实验室内部环境污染,更换引物探针等方法无法有效消除污染。因此在实验操作各环节均要严格规范,阳性对照要事先经过可靠灭活,并尽可能降低其使用浓度,扩增产物、实验废弃物需进行无害化处理,以避免引起实验室内部环境污染和其他生物安全事故。6.5 严格依法依规操作农业农村部于2020年8月27日发布《关于非洲猪瘟病毒诊断制品生产经营使用有关事宜的通知》(农办牧〔2020〕42号),要求:自2021年1月1日起,各有关部门和单位在动物检疫或疫病监测、诊断中,对生猪及其产品开展非洲猪瘟病毒检测,应当使用已取得我部核发的产品批准文号的非洲猪瘟病毒诊断制品,确保检测结果准确。按照此规定,实验室自行构建的非洲猪瘟检测方法将不能继续使用,必须改用获得兽药批准文号的商品化非洲猪瘟检测试剂盒。

北京临床基因扩增检验实验室筹建策划方案项目名称:2012年临床基因扩增检验实验室1 概述 临床基因扩增实验是专门用来检验艾滋病、乙型肝炎、禽疫病等病毒感染性疾病的一种检测手段。它可以通过将病毒体内所含的基因进行扩增的方法,测出一些病毒含量不高的感染者体内是否含有特定的病毒。由于该检测方法可以测出普通检验难以检测出的病毒并具有灵敏度高、特异性高、快捷、对样品要求低等优点,因此被临床医生广为认可,已广泛应用于医院的临床诊断和各防疫检测部门的禽疫病诊断。但是,这种实验需要有能保证绝对安全、配置合理的实验室和非常规范的操作为前提。近年来对临床基因扩增检验实验室的建设越来越得到重视,因为它对检测结果的可靠性、准确性和安全性起到至关重要的作用。本文主要从临床基因扩增检验实验室的平面布局,空调通风系统设计、气流控制和污染的防制几个方面对实验室设计中的主要特点进行了阐述。 临床基因扩增检验实验室设计的核心问题是如何避免污染。因此,实验室的平面布局、空调通风系统设计、气流控制等都是围绕这个核心问题进行的。下面就对这几个方面分别进说明。2 临床基因扩增实验室平面布局 临床基因扩增检验实验室原则上分为四个单独的工作区域:试剂贮存和准备区、标本制备区、扩增反应混合物配制和扩增区、扩增产物分析区。为避免交叉污染,进入各个工作区域必须严格遵循单一方向进行,即只能从试剂贮存和准备区→标本制备区→扩增反应混合物配制和扩增区→扩增产物分析区。 各实验区之间的试剂及样品传递应通过传递窗进行。临床基因扩增实验室平面布置示意图如图1所示。 http://ng1.17img.cn/bbsfiles/images/2011/11/201111250944_332855_2394712_3.jpg图1临床基因扩增实验室平面布置示意图3 实验室空调通风系统设计及压力控制 临床基因扩增实验室并没有严格的净化要求,但是为避免各个实验区域间交叉污染的可能性,宜采用全送全排的气流组织形式。同时,要严格控制送、排风的比例以保证各实验区的压力要求。3.1 试剂贮存和准备区 该实验区主要进行的操作为贮存试剂的制备、试剂的分装和主反应混合液的制备。试剂和用于标本制作的材料应直接运送至该区,不得经过其他区域。试剂原材料必须贮存在本区内,并在本区内制备成所需的贮存试剂。 对与气流压力的控制,本区并没有严格的要求。3.2 标本制备区 该区域主要进行的操作为临床标本的保存、核酸(RNA、DNA)提取、贮存及其加入至扩增反应管和测定RNA时cDNA的合成。 本区的压力梯度要求为:相对于邻近区域为正压,以避免从邻近区进入本区的气溶胶污染。另外,由于在加样操作中可能会发生气溶胶所致的污染,所以应避免在本区内不必要的走动。3.3 扩增反应混合物配制和扩增区 该区域主要进行的操作为DNA或cDNA扩增。此外,已制备的DNA模板和合成的cDNA(来自样本制备区)的加入和主反应混合液(来自试剂贮存和制备区)制备成反应混合液等也可在本区内进行。在巢式PCR测定中,通常在第一轮扩增后必须打开反应管,因此巢式扩增有较高的污染危险性,第二次加样必须在本区内进行。 本区的压力梯度要求为:相对于邻近区域为负压,以避免气溶胶从本区漏出。为避免气溶胶所致的污染,应尽量减少在本区内的不必要的走动。个别操作如加样等应在超净台内进行。3.4 扩增产物分析区 该区域主要进行的操作为扩增片段的测定。如使用全自动封闭分析仪器检测,此区域可不设。 本区是最主要的扩增产物污染来源,因此对本区的压力梯度的要求为:相对于邻近区域为负压,以避免扩增产物从本区扩散至其它区域。4 污染的预防与控制 临床基因扩增实验室设计的核心问题是如何避免污染。在实际工作中,常见的有以下几种污染类型:扩增产物的污染;天然基因组DNA的污染;试剂的污染以及标本间的污染。由于一旦发生污染,实验就必须停止,直到找到污染源为止,而且实验结果必须作废,需重新进行实验。所以发生污染后再围绕实验室来寻找污染源不但耗时而且繁琐,浪费人力物力。因此要避免污染,首先应是预防,而不是排除。4.1 工作区域的严格划分(1)各个实验区域设置合理;(2)各个实验区域要有明显的标记(如醒目的门牌或不同的地面颜色等),以避免各个不同实验区域设备物品、试剂等发生混淆。4.2 合理的系统设置(1)合理的空调通风系统设置,尽量采用全送全排的空调系统;(2)严格的气流压力控制,保证不同的实验区内不同的压力要求。[/fon
今天遇到个难解的困惑: 做某个基因荧光定量标曲的时候,感觉什么指标都很好了,如稀释度间的间距、重复性、溶解曲线、标准偏差值等等都基本满意了,但是扩增效率却怎么也上不去,重复了一次还是如此情况,跟前面其它几个基因的相比,大多数指标数据的并没有大的差异,但就是不知怎么今天做的这个基因扩增效率显示这么低。。。诚向各路高手请教,不知你们是否曾经遇到过类似的情况呢?可能的原因是什么呢?求帮忙分析下下。。谢谢各位了!
问题描述:核酸扩增体系如何判断是否受到抑制?解答:[align=left][font=宋体][color=black]聚合酶链式反应[/color][/font][font='Times New Roman','serif'][color=black]([url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url])[/color][/font][font=宋体][color=black]已经成为生物学研究中一项常用的技术,但核酸扩增核过程中会存在许多抑制因素。通过在反应体系中增加内参,可实时监控每一个反应孔内是否受到[/color][/font][font='Times New Roman','serif'][color=black][url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url][/color][/font][font=宋体][color=black]抑制或干扰,防止[/color][/font][font='Times New Roman','serif'][color=black][url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url][/color][/font][font=宋体][color=black]反应抑制剂引起的假阴性结果。[/color][/font][/align]以上内容来自仪器信息网《PCR实战宝典》
问题描述:扩增曲线常出现阳性对照有典型的“S”型扩增曲线,但不显示 Ct 值的原因及解决方案?以非洲猪瘟检测为例解答:[align=left][font=宋体][color=black]出现以上情况的主要原因是仪器默认的荧光阈值线过高[/color][/font][sup][font='Times New Roman','serif'][color=black][17][/color][/font][/sup][font=宋体][color=black]。[/color][/font][font=宋体][color=black]解决方案:一般将荧光阈值设在[/color][/font][font='Times New Roman','serif'][color=black] [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url] [/color][/font][font=宋体][color=black]指数扩增的起始,并高于阴性对照曲线。其数值选择与试剂盒的性能有关,对荧光强度较高的扩增曲线,阈值的设定不一定非要遵循固定的原则。[/color][/font][font=宋体][color=black]因此,针对试剂盒性能不同,实验室应建立自己的阈值范围,尽可能保证低值样本检测结果的稳定性[/color][/font][sup][font='Times New Roman','serif'][color=black][19][/color][/font][/sup][font=宋体][color=black]。[/color][/font][/align]以上内容来自仪器信息网《PCR实战宝典》
做PCR扩增做猪瘟检测??需要多哪些防护?望老师不吝赐教
问题描述:荧光定量[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url],扩增曲线有一向上或向下的尖峰的现象,可能是什么原因?解答:[align=left][font=宋体][color=black]([/color][/font][font='Times New Roman','serif'][color=black]1[/color][/font][font=宋体][color=black])反应过程中电压不稳定。[/color][/font][/align][align=left][font=宋体][color=black]([/color][/font][font='Times New Roman','serif'][color=black]2[/color][/font][font=宋体][color=black])可能在[/color][/font][font='Times New Roman','serif'][color=black] 20 [/color][/font][font=宋体][color=black]个循环左右时,仪器暂停了或被开盖,使光线突然增强。[/color][/font][/align][align=left][font=宋体][color=black]([/color][/font][font='Times New Roman','serif'][color=black]3[/color][/font][font=宋体][color=black])如果尖峰向下,可能是由卤素灯老化所致,这时应更换。[/color][/font][/align]以上内容来自仪器信息网《PCR实战宝典》
问题描述:荧光定量[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url],其中部分样本扩增效率过低如何改进?以诺如病毒检测为例解答:[align=left][font=宋体][color=black]可能的原因有:[/color][/font][/align][align=left][font=宋体][color=black]([/color][/font][font='Times New Roman','serif'][color=black]1[/color][/font][font=宋体][color=black])检查反应条件:如病毒中[/color][/font][font='Times New Roman','serif'][color=black][url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url][/color][/font][font=宋体][color=black]反应程序设定错误,将[/color][/font][font='Times New Roman','serif'][color=black] 42℃[/color][/font][font=宋体][color=black]设置成[/color][/font][font='Times New Roman','serif'][color=black] 50℃[/color][/font][font=宋体][color=black]。[/color][/font][/align][align=left][font=宋体][color=black]([/color][/font][font='Times New Roman','serif'][color=black]2[/color][/font][font=宋体][color=black])提取液残留,一定程度抑制了[/color][/font][font='Times New Roman','serif'][color=black] [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url] [/color][/font][font=宋体][color=black]反应。[/color][/font][/align][align=left][font=宋体][color=black]([/color][/font][font='Times New Roman','serif'][color=black]3[/color][/font][font=宋体][color=black])反应液未严格取量混匀或分装不均匀,建议规范操作,严格按照说明书操作。[/color][/font][/align]以上内容来自仪器信息网《PCR实战宝典》
问题描述:检测样品无典型“S”形扩增曲线,但其 Ct 值显示为阳性?以非洲猪瘟检测为例解答:[align=left][font=宋体][color=black]实际检测过程中,也会遇到被检样品无典型[/color][/font][font='Times New Roman','serif'][color=black]“S”[/color][/font][font=宋体][color=black]形扩增曲线,但其[/color][/font][font='Times New Roman','serif'][color=black] Ct [/color][/font][font=宋体][color=black]值显示为阳性(按照试剂盒说明书[/color][/font][font='Times New Roman','serif'][color=black] Ct [/color][/font][font=宋体][color=black]值判定标准)的情况。主要是由于被检样品不够新鲜或经反复冻融等,导致样本携带的病毒出现降解或样本核酸提取不纯。[/color][/font][sup][font='Times New Roman','serif'][color=black][18][/color][/font][/sup][font=宋体][color=black]。因此,被检样品要尽量新鲜,[/color][/font][font='Times New Roman','serif'][color=black]4℃~8℃[/color][/font][font=宋体][color=black]保存的血清或抗凝血样品一般不超过[/color][/font][font='Times New Roman','serif'][color=black] 120 h[/color][/font][font=宋体][color=black],唾液样品、组织样品冻融次数一般不超过[/color][/font][font='Times New Roman','serif'][color=black] 3 [/color][/font][font=宋体][color=black]次。[/color][/font][/align]以上内容来自仪器信息网《PCR实战宝典》

[font=arial, helvetica, sans-serif][color=#3333ff][b] [/b][/color][/font][align=center][font=arial, helvetica, sans-serif][b][size=24px]什么是荧光定量PCR[/size][/b][/font][/align][align=center][font=arial, helvetica, sans-serif][b][size=24px][/size][/b][/font][/align][font=arial, helvetica, sans-serif][color=#3333ff][color=#3333ff][b] [url=http://www.woyao17.com.cn/chanpinzhanshi/peitaoxiantiao/24.html]荧光定量PCR[/url][/b][/color][/color]又称qPCR,[color=#333333]是一种在DNA扩增反应中,以荧光化学物质测每次[/color]聚合酶链式反应[color=#333333]([/color][url=http://www.woyao17.com.cn/chanpinzhanshi/peitaoxiantiao/][color=#3333ff][b]PCR[/b][/color][/url][color=#333333])循环后产物总量的方法。通过内参或者外参法对待测样品中的特定DNA序列进行定量分析的方法。[/color][/font][size=16px][b][font=微软雅黑, sans-serif]荧光定量PCR的由来[/font][/b][/size][color=#333333] [/color][url=http://www.woyao17.com.cn][img=,690,460]https://ng1.17img.cn/bbsfiles/images/2020/09/202009270913440814_9803_4137464_3.jpg!w690x460.jpg[/img][/url] 2020年,一场突如其来的新冠疫情,让“[url=http://www.woyao17.com.cn/chanpinzhanshi/peitaoxiantiao/24.html][color=#0000ff][b]核酸检测[/b][/color][/url]”这一疾病检测方法一夜间为大家所公知。它在这次疫情防控中发挥了重大作用,成为临床诊断的 “金标准”。而实时荧光定量PCR则是此次核酸检测的主要应用方法,用以检测、判定临床样本中是否含有新冠病毒。今天小编先带大家初步了解一下什么是实时荧光定量PCR。 它的由来是这样的从20世纪90年代初开始,许多实验室开始致力于相对准确的定量PCR技术的研究。在这一过程中,应用较多的是半[url=http://www.woyao17.com.cn/chanpinzhanshi/peitaoxiantiao/24.html][color=#0000ff][b]定量PCR技术[/b][/color][/url]。引入管家基因,通过电泳后比较目的基因和管家基因的产物相对量,得到起始模板在量的差异。随后,为了进一步提高定量的准确性,人们开始设想在PCR过程中加入荧光物质,荧光物质会参与到第一轮PCR循环中,随着循环的进行和产物的增加,检测到的荧光物质也在发生变化,从而通过对荧光物质的检测实现全程监控PCR进程,最终可以计算出初始的模板量。1993年Higuchi等人将这一设想付诸实现!它的定义是这样的实时荧光定量PCR (Quantitative Real-time PCR)是一种在DNA扩增反应中,以荧光化学物质测每次聚合酶链式反应(PCR)循环后产物总量的方法。通过内参或者外参法对待测样品中的特定DNA序列进行定量分析的方法。由于在PCR扩增的指数时期,模板的Ct值和该模板的起始拷贝数存在线性关系,所以成为定量的依据。 [b][font=微软雅黑, sans-serif][size=16px]荧光定量PCR的原理[/size][/font][/b] 它检测的原理是这样的原理,包括探针类和非探针类两种。探针类是利用与靶序列特异杂交的探针来指示扩增产物的增加;非探针类则是利用荧光染料或者特殊设计的引物来指示扩增产物的增加。探针法由于增加了探针的识别步骤,特异性更高,而非探针类则简便易行。例如: 1.SYBRGreenⅠ法:SYBRGreenⅠ是一种结合于小沟中的双链DNA结合染料。在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。特点:非特异性,需检测熔解曲线;单通道检测;使用方便,经济便宜;常用于实验室研究。 2.TaqMan探针法:TaqMan探针是一种寡核苷酸探针,它的荧光与目的序列的扩增相关。它设计为与目标序列上游引物和下游引物之间的序列配对。探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物的形成完全同步。特点: 特异性好;可以多通道检测;单独设计探针;价格较高;常用于临床检测。随着荧光检测[url=http://www.woyao17.com.cn/chanpinzhanshi/peitaoxiantiao/24.html][color=#3333ff][b]PCR仪[/b][/color][/url]的商品化,实时荧光定量PCR进入了一个特异性强、灵敏度高、重复性好、定量准确、速度快、全封闭反应和由计算机软件进行快速统计的全新时代,实现了PCR从定性或半定量到准确定量的飞跃。它的特点和优势是这样的 1)特异性强:引物和探针的“双保险”,避免检测的假阳性。 2)灵敏度高:分析PCR产物的对数期,自动化仪器收集荧光信号,避免了许多人为因素干扰。 3)避免污染:全封闭反应,无须PCR后处理。 4)实现定量:运用标准品获得标准曲线,结合Ct值进行准确定量。 5)高效低耗:可实现一管多检。 6)操作简便:在线式实时监测扩增结果,不必接触有害物质。 7)快速:反应时间1.5小时。[size=16px][b][font=微软雅黑, sans-serif]在分子生物学领域的研究应用[/font][/b][/size][list=1][*]定量核酸浓度:传统方法是用琼脂糖凝胶电泳或者分光光度计测定核酸浓度,其结果不准确而且易污染,实时荧光定量PCR可以解决这些问题,它的准确性、灵敏度高且无污染,对一些传染性疾病进行定量分析、病原微生物和病毒含量检测,此项技术都是首选 。[*]研究基因表达:应用实时定量PCR技术可以对基因时间、空间表达水平差异进行比较。例如,对特定基因用物理、化学、药物等不同方法处理后的差异进行比较,为人们的科学研究提供依据 。[*]用于单核苷酸多态性(SNP)检测分析:人们对疾病的易感性和对同一种药物治疗同一种疾病的效果是有差异性的,遗传物质DNA的多态性RELP,STR,ABO血型和SNP是个体差异的遗传基础。SNP在人类基因组中广泛存在,是人类可遗传变异中最常见的一种,在遗传性疾病的研究中具有重要意义 。[*]DNA甲基化检测:DNA甲基化是表观遗传学重要的标记信息,通过实时荧光定量PCR技术获得基因组甲基化水平数据对表观遗传学的时空特异性研究具有重要意义。[/list][url=http://www.woyao17.com.cn/chanpinzhanshi/][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2020/09/202009270916423385_1359_4137464_3.jpg!w690x517.jpg[/img][/url]
【仪器心得】实时荧光定量PCR仪使用心得 在生物科学研究领域,实时荧光定量PCR(Quantitative Real-Time PCR,简称qPCR)技术已经成为不可或缺的一部分。这一技术不仅具备高灵敏度、高特异性和高精度的特点,还能够对DNA或RNA进行定量分析,为基因表达分析、病原体检测、遗传病诊断以及药物研发等领域提供了强大的支持。作为一名分子生物学研究者,我对于实时荧光定量PCR仪的使用有着深刻的心得体会,以下将结合技术原理、特点、应用场景以及个人见解,为大家详细介绍这一强大的科研工具。 https://ng1.17img.cn/bbsfiles/images/2024/10/202410121429543053_6906_6748942_3.jpeg实时荧光定量PCR仪技术详解原理 实时荧光定量PCR技术基于PCR(聚合酶链式反应)的基本原理,通过不断循环的变性、退火和延伸步骤,实现DNA或RNA的指数级扩增。与常规PCR不同的是,实时荧光定量PCR在反应体系中加入了荧光染料或荧光探针,这些荧光物质能够与扩增产物特异性结合,并在特定波长光的激发下发出荧光。随着PCR反应的进行,荧光信号逐渐增强,仪器通过实时监测荧光信号的变化,可以绘制出扩增曲线,进而对样本中的目标核酸进行定量分析。 特点 1. 高灵敏度:实时荧光定量PCR技术能够检测到极微量的目标核酸,使得其在低丰度基因表达分析、病原体微量检测等方面具有显著优势。 2. 高特异性:通过设计特异性的引物和探针,实时荧光定量PCR可以实现对目标基因的精确识别,避免了非特异性扩增的干扰。 3. 高精度:结合先进的荧光信号检测技术和数据分析算法,实时荧光定量PCR能够提供准确的定量结果,有助于科研工作者进行精确的数据分析和比较。 4. 快速便捷:现代实时荧光定量PCR仪通常配备有高效的热循环系统和自动化的样品处理功能,大大缩短了实验时间,提高了工作效率。 应用场景 实时荧光定量PCR技术在生命科学领域的应用广泛而深入。在基因表达分析中,它可以用于检测不同组织、不同发育阶段或不同处理条件下基因的表达水平变化;在病原体检测中,它能够快速准确地识别并定量病原体核酸,为疾病诊断和治疗提供重要依据;在遗传病诊断中,实时荧光定量PCR可以检测基因突变、基因缺失或基因重复等异常情况;此外,在药物研发、环境监测和食品安全等领域,实时荧光定量PCR技术也发挥着重要作用。 个人使用心得与推荐 作为一名长期使用实时荧光定量PCR仪的科研人员,我深刻体会到了这一技术带来的便利和优势。在使用过程中,我注意到以下几点对于获得准确可靠的实验结果至关重要: 1. 样本处理:样本的纯净度和完整性对于实验结果至关重要。因此,在进行实验前,需要对样本进行严格的预处理和质量控制,以确保样本中的目标核酸不受污染和降解。 2. 引物和探针设计[size=14px]:引物和探针的特异性是实时荧光定量PCR技术成功的关键。在设计引物和探针时,需要充分考虑目标基因的序列特征、潜在的二级结构以及可能的非特异性扩增等因素,以确保实验的准确性和可靠性。 3. 仪器选择和校准:不同品牌和型号的实时荧光定量PCR仪在性能上可能存在差异。因此,在选择仪器时,需要根据实验需求和预算进行综合考虑。同时,定期对仪器进行校准和维护也是确保实验结果准确性的重要措施。 4. 在我个人的使用体验中,某品牌的实时荧光定量PCR仪给我留下了深刻印象。该仪器不仅具备高效的热循环系统和精准的荧光信号检测系统,还配备了智能化的操作界面和数据分析软件,使得实验操作更加便捷、数据分析更加准确。此外,该仪器还支持多种荧光通道和反应体系的选择,能够满足不同实验需求。 https://ng1.17img.cn/bbsfiles/images/2024/10/202410121429546220_2116_6748942_3.jpeg结语 实时荧光定量PCR技术作为生命科学领域的重要工具之一,其高精度、高灵敏度和高特异性的特点使得它在众多研究领域都发挥着不可替代的作用。通过深入了解实时荧光定量PCR仪的原理、特点和应用场景,并结合个人使用心得和推荐,相信广大科研工作者能够更好地利用这一技术,探索生命科学的奥秘,推动科研事业的进步和发展。
最近网上传有“进入食品安全监测领域的微流控恒温扩增仪”这个仪器是啥?能测定什么项目?
荧光定量PCR简介荧光定量PCR检测技术诞生至今已10多年的时间,而其应用一直都没广泛展开,究其原因,无外乎受制于相关仪器、试剂和技术的发展。近期,尤其是08年以来,仪器和试剂是遍地开花,这也使科研人员均跃跃欲试,都想借此技术使自己的研究能突飞猛进,发展势头通过查找每年所发表的文章数可一目了然。据有关统计,在 Medline 数据库中,用“Taqman” 或 ”real time PCR” 作为关键词检索,1996 年是19 篇,1999 年157 篇,到2003 年就高达2984 篇,2009年会是多少呢?我们不得而知。但其迅猛的发展势头却是不可更改的,本公司真诚希望能和众多研究人员共同努力,抓住这一大好时机,为科研事业的发展贡献自身的力量。基于这一目标,本公司长期以来对荧光定量PCR,无论是技术还是多年来的产品,均进行了深入地研究,并及时推出更加优异的荧光定量 PCR技术服务。荧光定量PCR原理荧光定量PCR最早称TaqMan PCR,后来也叫Real-Time PCR,是美国PE(Perkin Elmer)公司1995年研制出来的一种新的核酸定量技术。该技术是在常规PCR基础上加入荧光标记探针或相应的荧光染料来实现其定量功能的。其原理:随着PCR反应的进行,PCR反应产物不断累计,荧光信号强度也等比例增加。每经过一个循环,收集一个荧光强度信号,这样我们就可以通过荧光强度变化监测产物量的变化,从而得到一条荧光扩增曲线图。一般而言,荧光扩增曲线可以分成三个阶段:荧光背景信号阶段,荧光信号指数扩增阶段和平台期。在荧光背景信号阶段,扩增的荧光信号被荧光背景信号所掩盖,无法判断产物量的变化。而在平台期,扩增产物已不再呈指数级的增加,PCR 的终产物量与起始模板量之间没有线性关系,根据最终的 PCR 产物量也不能计算出起始 DNA 拷贝数。只有在荧光信号指数扩增阶段, PCR产物量的对数值与起始模板量之间存在线性关系,我们可以选择在这个阶段进行定量分析。为了定量和比较的方便,在实时荧光定量 PCR 技术中引入了两个非常重要的概念:荧光阈值和 CT值(如下图所示)。荧光域值(threshold)是在荧光扩增曲线上人为设定的一个值,它可以设定在荧光信号指数扩增阶段任意位置上,但一般荧光域值的缺省设置是PCR反应前3-15个循环荧光信号标准偏差的10倍,即:threshold。http://www.biomart.cn/upload/asset/2011/01/25/1295364674.jpgCt 值:是指每个反应管内的荧光信号到达设定域值时所经历的循环数。Ct值与起始模板的关系:研究表明,每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系,起始拷贝数越多,Ct值越小。利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代表Ct值如下图所示。因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。http://www.biomart.cn/upload/asset/2011/01/25/1295364675.jpg荧光定量检测荧光定量检测根据所使用的标记物不同可分为荧光探针和荧光染料。荧光探针又包括Beacon技术(分子信标技术,以美国人Tagyi为代表)、 TaqMan探针(以美国ABI公司为代表)和FRET技术(以罗氏公司为代表)等;荧光染料包括饱和荧光染料和非饱和荧光染料,非饱和荧光染料的典型代表就是现在最常用的SYBR GreenⅠ;饱和荧光染料有EvaGreen、LC Green等。嵌合荧光染料法(SYBR GreenⅠ)SYBR Green I是荧光定量PCR最常用的DNA结合染料,与双链DNA非特异性结合。在游离状态下,SYBR Green I发出微弱的荧光,但一旦与双链DNA结合,其荧光增加1000倍。所以,一个反应发出的全部荧光信号与出线的双链DNA量呈比列,且会随扩增产物的增加而增加。http://www.biomart.cn/upload/asset/2011/01/25/1295364672.gifSYBR Green I荧光染料与DNA双链的结合双链DNA结合染料的优点:实验设计简单,仅需要2个引物,不需要设计探针,无需设计多个探针即可以快速检验多个基因,且能够进行熔点曲线分析,检验扩增反应的特异性,低的初始成本,通用性好,因此国内外在科研中使用比较普遍。荧光探针法(Taqman 技术):PCR扩增时,加入一对引物的同时再加入一个特异性的荧光探针。该探针为一直线型的寡核苷酸,两端分别标记一个荧光报告基团和一个荧光淬灭基团,探针完整时,报告基团发射的荧光信号被淬灭基团吸收,PCR 仪检测不到荧光信号; PCR扩增时(在延伸阶段),Taq 酶的 5' - 3' 切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条 DNA 链,就有一个荧光分子形成,实现了荧光信号的累积与 PCR 产物形成完全同步,这也是定量的基础所在。其过程如下图所示http://www.biomart.cn/upload/asset/2011/01/25/1295364673.gif荧光定量PCR的应用分子生物学研究1、核酸定量分析。 对传染性疾病进行定量定性分析,病原微生物或病毒含量的检测 , 比如近期流行的甲型H1N1流感, 转基因动植物基因拷贝数的检测,RNAi 基因失活率的检测等。2 、基因表达差异分析。 比较经过不同处理样本之间特定基因的表达差异 ( 如药物处理、物理处理、化学处理等 ) ,特定基因在不同时相的表达差异以及cDNA芯片或差显结果的确证3 、SNP 检测。检测单核苷酸多态性对于研究个体对不同疾病的易感性或者个体对特定药物的不同反应有着重要的意义,因分子信标结构的巧妙性,一旦SNP 的序列信息是已知的,采用这种技术进行高通量的 SNP 检测将会变得简单而准确。4、 甲基化检测。甲基化同人类的许多疾病有关,特别是癌症, Laird 报道了一种称作 Methylight的技术,在扩增之前先处理 DNA ,使得未甲基化的胞嘧啶变成尿嘧啶,而甲基化的胞嘧啶不受影响,用特异性的引物和 Taqman探针来区分甲基化和非甲基化的 DNA ,这种方法不仅方便而且灵敏度更高。医学研究1、 产前诊断:人们对遗传性物质改变引起的遗传性疾病还无法治疗,到目前为止,还只能只能通过产前监测,减少病婴出生,以防止各类遗传性疾病的发生,如为减少X连锁遗传病患儿的出生,从孕妇的外周血中分离胎儿DNA,用实时荧光定量PCR检测其Y性别决定区基因是一种无创伤性的方法,易为孕妇所接受。2、 病原体检测:采用荧光定量PCR检测技术可以对淋球菌、沙眼衣原体、解脲支原体、人类乳头瘤病毒、单纯疱疹病毒、人类免疫缺陷病毒、肝炎病毒、流感病毒、结核分枝杆菌、EB病毒和巨细胞病毒等病原体进行定量测定。与传统的检测方法相比具有灵敏度高、取样少、快速简便等优点。3、 药物疗效考核:对乙型肝炎病毒 (HBV)、丙型肝炎病毒 (HCV) 定量分析显示:病毒量与某些药物的疗效关系。HCV高水平表达,干扰素治疗作用不敏感,而HCV低滴度,干扰素作用敏感;在拉米夫定治疗过程中,HBV- DNA的血清含量曾有下降,随后若再度上升或超出以前水平,则提示病毒发生变异。4、 肿瘤基因检测:尽管肿瘤发病的机理尚未清楚,但相关基因发生突变是致癌性转变的根本原因已被广泛接受。癌基因的表达增加和突变,在许多肿瘤早期就可以出现。实时荧光定量 PCR不但能有效地检测到基因的突变,而且可以准确检测癌基因的表达量。目前用此方法进行过端粒酶hTERT基因、慢性粒细胞性白血病WT1基因、肿瘤 ER基因、前列腺癌PSM基因、肿瘤相关的病毒基因等多种基因的表达检测。
问题1:不出现扩增条带 引物:引物质量是PCR失败或扩增条带不理想、容易弥散的常见原因。对策为:①选定合适的引物合成单位;②引物的浓度不仅要看OD值,最好用引物原液做琼脂糖凝胶电泳,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应与引物合成单位协商解决,如一条引物亮度高,一条引物亮度低,在稀释引物时要平衡其浓度;③引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏而导致变质降解失效;④引物设计不合理,如引物长度不够,引物之间形成二聚体等。 Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。 靶序列变异:如靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。问题2:出现非特异性扩增带 PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。非特异性条带出现的原因:一是引物与靶序列不完全互补,或引物聚合形成二聚体;二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。 其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。对策为:必要时重新设计引物;减低酶量或调换另一来源的酶;降低引物量,适当增加模板量,减少循环次数;适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。问题3:出现片状拖带或涂抹带 PCR扩增有时出现涂抹带或片状带或地毯样带。可能由于Mg2+浓度过高,退火温度过低,循环次数过多,GC含量过高引起。对策为:适当降低Mg2+浓度;增加模板量;减少循环次数;加入添加剂甘油或Qiagen公司的Q-solution。问题4:污染的监测——对照试验 1.阳性对照:应设有阳性对照,它是PCR反应是否成功、产物条带位置及大小是否合乎理论要求的一个重要标志。阳性对照要选择扩增度中等、重复性好,经各种鉴定是该产物的标本,如以重组质粒为阳性对照,其含量宜低不宜高(100个拷贝以下)。 2.阴性对照:每次PCR实验务必做阴性对照。它包括①标本对照:被检的标本是血清就用鉴定后的正常血清作对照,被检的标本是组织细胞就用相应的组织细胞作对照;②试剂对照:在PCR试剂中不加模板DNA或RNA进行PCR扩增,以监测试剂是否污染。 3.重复性试验 4.选择不同区域的引物进行PCR扩增问题5:污染的处理 (一)环境污染 1.稀酸处理法 2.紫外照射(UV)法 (二)反应液污染,可采用下列方法之一处理: 1.DNase I法 2.内切酶法 3.紫外照射法 4.Gama射线辐射法 (三)尿嘧啶糖苷酶(UNG)法 由于UV照射的去污染作用对500bp以下的片段效果不好,而临床用于检测的PCR扩增片段通常为300bp左右,因此UNG的预防作用日益受到重视和肯定。 (四)固相捕获法 用于去除标本中污染的核酸和杂质。 (五)RS-PCR法(RNA-specificPCR) 也称为链特异性PCR,主要指用于RNA模板的特异性PCR法,该法可明显降低假阳性而不影响PCR的敏感性。 (六)抗污染引物法 该对引物扩增时通过病毒DNA克隆如入质粒的位点,这一区域只存在于完整的原病毒中。如果重组质粒污染了标本,就不能扩增出任何条带,即使出现了扩增带,其大小也与预期的不同。只有原病毒DNA才能被引物扩增,因此只要出现预期大小的扩增带就可以证明标本是阳性的,该法试用于环状靶分子系列。
什么是PCR技术? PCR(Polymerase Chain Reaction)技术,中文译为聚合酶链式反应,是一种在体外(试管、切片…)扩增核酸的技术。PCR的基本反应包括以DNA为模板的反应和以mRNA为模板的反应。 PCR技术是模拟体内天然DNA(脱氧核糖核酸)的复制过程。以扩增DNA为例,其基本原理是在模板、引物、4种dNTP和耐热DNA聚合酶存在的条件下,特异扩增位于两段已知序列之间的DNA区段的酶促合成反应。
PCR相关经验分享PCR常见问题-分析及对策(无扩增产物、非特异性扩增、拖尾、假阳性)问题1:无扩增产物 现象:正对照有条带,而样品则无 原因: 1.模板:含有抑制物,含量低 2.Buffer对样品不合适 3.引物设计不当或者发生降解 4.反应条件:退火温度太高,延伸时间太短 对策: 1.纯化模板或者使用试剂盒提取模板DNA或加大模板的用量 2.更换Buffer或调整浓度 3.重新设计引物(避免链间二聚体和链内二级结构)或者换一管新引物 4.降低退火温度、延长延伸时间 问题2:非特异性扩增 现象:条带与预计的大小不一致或者非特异性扩增带 原因: 1.引物特异性差 2.模板或引物浓度过高 3.酶量过多 4.Mg2+浓度偏高 5.退火温度偏低 6.循环次数过多 对策: 1.重新设计引物或者使用巢式PCR 2.适当降低模板或引物浓度 3.适当减少酶量 4.降低镁离子浓度 5.适当提高退火温度或使用二阶段温度法 6.减少循环次数 问题3:拖尾 现象:产物在凝胶上呈Smear状态。 原因: 1.模板不纯 2.Buffer不合适 3.退火温度偏低 4.酶量过多 5.dNTP、Mg 2+浓度偏高 6.循环次数过多 对策: 1.纯化模板 2.更换Buffer 3.适当提高退火温度 4.适量用酶 5.适当降低dNTP和镁离子的浓度 6.减少循环次数 问题4:假阳性 现象:空白对照出现目的扩增产物 原因: 靶序列或扩增产物 的交*污染 对策: 1.操作时应小心轻柔,防止将靶序列吸入加样枪内或溅出离心管外; 2.除酶及不能耐高温的物质外,所有试剂或器材均应高压消毒。所用离心管 及加样枪头等均应一次性使用。 3.各种试剂最好先进行分装,然后低温贮存 PCR引物设计的黄金法则 (转自tiangen)1.引物最好在模板cDNA的保守区内设计。 DNA序列的保守区是通过物种间相似序列的比较确定的。在NCBI上搜索不同物种的同一基因,通过序列分析软件(比如DNAman)比对(Alignment),各基因相同的序列就是该基因的保守区2.引物长度一般在15~30碱基之间。 引物长度(primer length)常用的是18-27 bp,但不应大于38,因为过长会导致其延伸温度大于74℃,不适于Taq DNA 聚合酶进行反应。3.引物GC含量在40%~60%之间,Tm值最好接近72℃。 GC含量(composition)过高或过低都不利于引发反应。上下游引物的GC含量不能相差太大。另外,上下游引物的Tm值(melting temperature)是寡核苷酸的解链温度,即在一定盐浓度条件下,50%寡核苷酸双链解链的温度。有效启动温度,一般高于Tm值5~10℃。若按公式Tm= 4(G+C)+2(A+T)估计引物的Tm值,则有效引物的Tm为55~80℃,其Tm值最好接近72℃以使复性条件最佳。4.引物3′端要避开密码子的第3位。 如扩增编码区域,引物3′端不要终止于密码子的第3位,因密码子的第3位易发生简并,会影响扩增的特异性与效率。5.引物3′端不能选择A,最好选择T。 引物3′端错配时,不同碱基引发效率存在着很大的差异,当末位的碱基为A时,即使在错配的情况下,也能有引发链的合成,而当末位链为T时,错配的引发效率大大降低,G、C错配的引发效率介于A、T之间,所以3′端最好选择T。6. 碱基要随机分布。 引物序列在模板内应当没有相似性较高,尤其是3’端相似性较高的序列,否则容易导致错误引发(False priming)。降低引物与模板相似性的一种方法是,引物中四种碱基的分布最好是随机的,不要有聚嘌呤或聚嘧啶的存在。尤其3′端不应超过3个连续的G或C,因这样会使引物在GC富集序列区错误引发。7. 引物自身及引物之间不应存在互补序列。 引物自身不应存在互补序列,否则引物自身会折叠成发夹结构(Hairpin)使引物本身复性。这种二级结构会因空间位阻而影响引物与模板的复性结合。引物自身不能有连续4个碱基的互补。 两引物之间也不应具有互补性,尤其应避免3′ 端的互补重叠以防止引物二聚体(Dimer与Cross dimer)的形成。引物之间不能有连续4个碱基的互补。 引物二聚体及发夹结构如果不可避免的话,应尽量使其△G值不要过高(应小于4.5kcal/mol)。否则易导致产生引物二聚体带,并且降低引物有效浓度而使PCR 反应不能正常进行。8. 引物5′ 端和中间△G值应该相对较高,而3′ 端△G值较低。 △G值是指DNA 双链形成所需的自由能,它反映了双链结构内部碱基对的相对稳定性,△G值越大,则双链越稳定。应当选用5′ 端和中间△G值相对较高,而3′ 端△G值较低(绝对值不超过9)的引物。引物3′ 端的△G 值过高,容易在错配位点形成双链结构并引发DNA 聚合反应。(不同位置的△G值可以用Oligo 6软件进行分析)9.引物的5′端可以修饰,而3′端不可修饰。 引物的5′ 端决定着PCR产物的长度,它对扩增特异性影响不大。因此,可以被修饰而不影响扩增的特异性。引物5′ 端修饰包括:加酶切位点;标记生物素、荧光、地高辛、Eu3+等;引入蛋白质结合DNA序列;引入点突变、插入突变、缺失突变序列;引入启动子序列等。 引物的延伸是从3′ 端开始的,不能进行任何修饰。3′ 端也不能有形成任何二级结构可能。10. 扩增产物的单链不能形成二级结构。 某些引物无效的主要原因是扩增产物单链二级结构的影响,选择扩增片段时最好避开二级结构区域。用有关软件(比如RNAstructure)可以预测估计mRNA的稳定二级结构,有助于选择模板。实验表明,待扩区域自由能(△G°)小于
荧光定量PCR(也称TaqMan PCR,以下简称FQ-PCR)是美国PE(Perkin Elmer)公司1995年研制出来的一种新的核酸定量技术,该技术是在常规PCR基础上加入荧光标记探针来实现其定量功能的,与变通PCR相比,FQ-PCR具有许多优点。本文拟就该技术的特点、原理和方法以及应用作一简要叙述。一、特点FQ-PCR不仅具有普通PCR的高灵敏性,而且由于荧光探针的应用,可以通过光电传导系统直接探测PCR扩增过程中荧光信号的变化以获得定量结果,所以还具有DNA杂交的高特异性和光谱技术的高精确性,克服了常规PCR的许多缺点。如一般PCR产物都需通过琼脂糖凝胶电泳和溴化乙锭染色紫外光观察结果或通过聚丙烯酰胺凝胶电泳和银染检测,不仅需要多种仪器,而且费时费力,所使用的染色剂溴化乙锭对人体又有害,这些繁杂的实验过程又给污染和假阳性提供了机会。而FQ-PCR只须在加样时打开一次盖子,其后的过程完全是闭管操作,不需要PCR后处理,避免了常规PCR操作中的诸多弊端。实验一般使用PE公司研制的ABI7100型PCR扩增仪。该仪器具有以下特点:①应用广泛:可用于DNA和RNA的PCR产物定量、基因表达研究、病原体检测及PCR条件的优化等。②独特的定量原理:采用荧光标记探针,经激光激发后荧光量随PCR循环而累积,从而达到定量目的。③工作效率高:内置9600型PCR扩增仪,电脑控制1~2小时全自动同步完成96个样品的扩增及定量。④无须凝胶电泳:无须对样品进行稀释和电泳,只须通过特殊探头在反应管内直接检测。⑤无管道内污染:采用独特全封闭反应管及光电传导系统,无须顾及污染。⑥结果重现性好:定量动态范围高达五个数量级。所以自从此项技术研制成功以来,受到许多科研工作者的重视并在多个领域得到应用。二、原理和方法FQ-PCR的工作原理是利用Taq酶的5’→3’外切酶活性,在PCR反应系统中加入一个荧光标记探针。该探针可与引物包含序列内的DNA模板发生特异性杂交,探针的5’端标以荧光发射基因FAM(6-羧基荧光素,荧光发射峰值在518nm处),靠近3’端标以荧光淬灭基团TAMRA(6-羧基四甲基若丹明,荧光发射峰值在582nm处),探针的3’开端被磷酸化以防止探针在PCR扩增过程中被延伸。当探针保持完整时,淬灭基团抑制发射基团的荧光发射。发射基团一旦与淬灭基团发生分离,抑制作用被解除,518nm处的光密度增加而被荧光探测系统检测到。复性期探针与模板DNA发生杂交,延伸期Taq酶随引物延伸沿DNA模板移动,当移动到探针切断,淬灭作用被解除,荧光信号释放出来(见图)。模板每复制一次,就有一个探针被切断,伴随一个荧光信号的释放。由于被释放的荧光基团数目和PCR产物数量是一对一的关系,因此用该技术可对模板进行准确定量。实验仪器一般使用PE公司研制的ABI7100型 PCR扩增仪,也可用其它PCR仪。如果用ABI7700型反应型反应系统进行实验,反应结束后,通过电脑分析,可直接给出定量结果。如果用其他PCR仪,则需要同时使用荧光探测仪测量反应管中的荧光信号,计算出RQ+、RQ-、△RQ。RQ+代表样品管荧光发射基团发光强度与淬灭基团发光强度的比率,RQ-代表空白管中二者的比率,△RQ(△RQ=RQ+-RQ-)代表PCR过程中荧光信号变化量,经过数据处理,即可得出定量结果。由于荧光探针的引入,显著提高了实验的特异性。探针设计一般应符合以下条件:①探针长度应在20~40个碱基左右,以保证结合的特异性。②GC碱基含量在40%~60%,避免单核苷酸序列的重复。③避免与引物发生杂交或重叠。④探针与模板结合的稳定程度要大于引物与模板结合的稳定程度,因此探针的Tm值要比引物的Tm值至少高出5℃。另外,探针的浓度、探针与模板序列的同源性,探针与引物的距离都对实验结果有影响。 http://www.biomart.cn/upload/asset/2008/07/21/1216476450.gif图.TaqMan PCR反应模式 (A)聚合反应;(B)链置换;(C)裂解; (D)聚合完成(R:FAM;Q:TAMRA,FP:上游引物;RP:下游引物)
急!急!今天进行PCR扩增,为什么没有荧光反应?是染料过期还是什么问题?
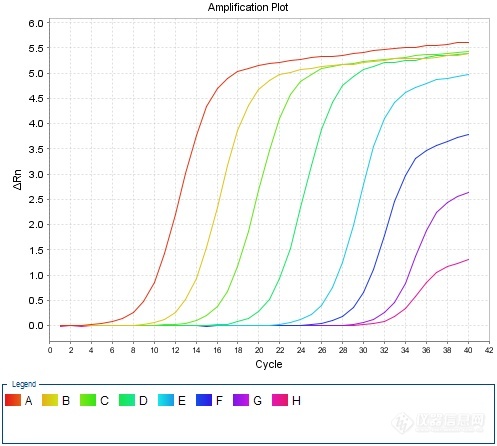
动物A型流感荧光定量[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]检测方法的模块化构建及实践1 前言流感病毒(influenza virus)属正粘病毒科,根据抗原特性及其基因特性的不同,流感病毒分为甲型(A型)、乙型(B型)、丙型(C型)。乙型变异性较弱、丙型抗原性比较稳定,仅感染人类;甲型(A型)抗原变异性最强,感染人类和其他动物,常引起世界性大流行。甲型流感病毒中至今发现能直接感染人的禽流感病毒亚型有:甲型H1N1、H5N1、H7N1、H7N2、H7N3、H7N7、H7N9、H9N2和H10N8。其中H1、H5、H7亚型为高致病性,H1N1、H5N1、H7N9尤为值得关注。甲型流感病毒根据H和N抗原不同,又分为许多亚型,H可分为18个亚型(H1~H18),N有11个亚型(N1~N11)。其中仅H1N1、H2N2、H3N2主要感染人类,其它许多亚型的自然宿主是多种禽类和动物。其中对禽类危害最大的为H5、H7和H9亚型毒株。一般情况下,禽流感病毒不会感染鸟类和猪以外的动物。但1997年香港首次报道发生18例H5N1人禽流感感染病例,其中6例死亡,引起全球广泛关注。1997年以后,世界上又先后几次发生了禽流感病毒感染人的事件。具有高致病性的H5N1、H7N7、H7N9、H9N2等禽流感病毒,一旦发生变异而具有人与人的传播能力,会导致人间禽流感流行,预示着禽流感病毒对人类已具有很大的潜在威胁。为确保公共卫生安全,以及畜牧业健康发展,有必要实施A型动物流感核酸监测,尽早发现疫情隐患并予以及时处置,避免重大动物疫情及公共卫生安全事件发生。2 模块化检测方法思路总体思路是:将动物疫病荧光定量[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]检测分为三个模块,核酸提取采用DNA/RNA共提试剂,核酸扩增采用预混液+引物探针方式,质控品利用抗原或商品化疫苗自制。具体思路如下:——核酸提取采用DNA/RNA共提试剂;——引物探针严格按照标准方法,委托生物工程公司合成;——反应体系中其他成分,采购商品化RT-q[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]预混液(含逆转录酶);——质控品:阳性对照采用病毒抗原、商品化疫苗等,测定结果后按一定比例稀释,保存备用。——反应体系:设为20μL,其中包含:预混液(2×)10μL、逆转录酶1μL、ROX 0.4μL为固定量,引物探针加入量根据检测标准中的终浓度予以换算,加灭菌水补足18μL,DNA或RNA模板为2μL;——反应条件首先按照标准中的条件开展摸索,如达到预期效果则予以固定;如效果不佳则参照预混液及引物探针推荐条件进行适当调整。3 材料和方法3.1 病毒禽流感血凝抑制抗原,包括H5亚型Re-4、Re-6、Re-7、Re-8、Re-11、Re-12等毒株,以及H7、H7N9、H7N9Re-2等毒株。3.2 试剂DNA/RNA共提试剂盒:FastPure Viral DNA/RNA Mini Kit试剂盒,南京诺维赞生产;RT-q[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]预混液:2× One Step Q Probe Mix,南京诺维赞生产;逆转录酶:One Step Q Probe Enzyme Mix,南京诺维赞生产。3.3 引物探针按照GB/T27539-2011《动物流感检测 A型流感病毒通用荧光RT-[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]检测方法》提供的序列,委托上海生工合成,引物稀释成10μmol/L、探针稀释成5μmol/L,分装成小包装冷冻保存备用。具体序列如下;正向引物:5’-GTC TTC TAA CCG AGG TCG AAA C-3’反向引物:5’-AAG ATC TGT GTT CTT TCC T[url=https://insevent.instrument.com.cn/t/Mp]gc[/url] AAA-3’探 针:5’-FAM-CCC TCA AAG CCG AGA TCG C-TAMRA-3’3.4 反应体系根据GB/T27539-2011所载反应体系,按照终浓度一致原则予以转化。具体转换方法:——反应体系总体积设定为20μL;——将标准中反应体系的10×[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url] Buffer、M[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]l2、dNTPs、Taq酶、反转录酶等成分转换为RT-q[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]预混液10μL、逆转录酶1μL(推荐量);——因使用ABI7500荧光[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]仪,故在体系中加入ROX 0.4μL;——标准中正向引物、反向引物、探针的终浓度分别为0.2μmol/L、0.2μmol/L和0.16μmol/L,故在修改后的体系中加入量为0.4μL、0.4μL和0.64μL;——总RNA加入量修改为2μL(根据经验该加入量已经足够);——根据上述各成分用量,补充体积的无核酸酶水修改为5.16μL。[table][tr][td=2,1][align=center][color=black]GB/T27539-2011[/color][/align][/td][td] [/td][td=2,1][align=center][color=black]修改方法[/color][/align][/td][/tr][tr][td][color=black]10× [url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url] Buffer[/color][/td][td][align=center][color=black]1.25[/color][/align][/td][td] [/td][td][color=black]2× RT-q[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]预混液[/color][/td][td][align=center][color=black]10[/color][/align][/td][/tr][tr][td][color=black]正向引物(10μmol/L)[/color][/td][td][align=center][color=black]0.5[/color][/align][/td][td] [/td][td][color=black]酶混合液[/color][/td][td][align=center][color=black]1[/color][/align][/td][/tr][tr][td][color=black]反向引物(10μmol/L)[/color][/td][td][align=center][color=black]0.5[/color][/align][/td][td] [/td][td][color=black]ROX[/color][/td][td][align=center][color=black]0.4[/color][/align][/td][/tr][tr][td][color=black]探针(5μmol/L)[/color][/td][td][align=center][color=black]0.8[/color][/align][/td][td] [/td][td][color=black]正向引物(10μmol/L)[/color][/td][td][align=center][color=black]0.4[/color][/align][/td][/tr][tr][td][color=black]M[url=https://insevent.instrument.com.cn/t/Mp]gc[/url]l2[/color][/td][td][align=center][color=black]0.75[/color][/align][/td][td] [/td][td][color=black]反向引物(10μmol/L)[/color][/td][td][align=center][color=black]0.4[/color][/align][/td][/tr][tr][td][color=black]dNTPs[/color][/td][td][align=center][color=black]0.1[/color][/align][/td][td] [/td][td][color=black]探针(5μmol/L)[/color][/td][td][align=center][color=black]0.64[/color][/align][/td][/tr][tr][td][color=black]Taq酶(5U/μL)[/color][/td][td][align=center][color=black]0.25[/color][/align][/td][td] [/td][td][color=black]总RNA[/color][/td][td][align=center][color=black]2[/color][/align][/td][/tr][tr][td][color=black]反转录酶[/color][/td][td][align=center][color=black]0.25[/color][/align][/td][td] [/td][td][color=black]无核酸酶水[/color][/td][td][align=center][color=black]5.16[/color][/align][/td][/tr][tr][td][color=black]无核酸酶水[/color][/td][td][align=center][color=black]10.6[/color][/align][/td][td] [/td][td][color=black]总体积[/color][/td][td][align=center][color=black]20[/color][/align][/td][/tr][tr][td][color=black]总RNA[/color][/td][td][align=center][color=black]10[/color][/align][/td][td] [/td][td] [/td][td] [/td][/tr][tr][td][color=black]总体积[/color][/td][td][align=center][color=black]25[/color][/align][/td][td] [/td][td] [/td][td] [/td][/tr][/table]3.5 反应条件参照GB/T27539-2011推荐的扩增程序,结合ABI7500荧光采集时间不能少于34s的要求,进行预试验。扩增程序如下:42℃30min→92℃3min→(92℃10s→45℃30s→72℃1min)*5→(92℃10s→60℃34s)*40如扩增结果理想,则不做修改直接采纳,否则进行适当调整。4 试验结果对禽流感病毒包括H5亚型Re-4、Re-6、Re-7、Re-8、Re-11、Re-12等毒株,以及H7、H7N9、H7N9Re-2等毒株均进行了核酸检测,结果均较理想。部分扩增曲线如下:[img]https://ng1.17img.cn/bbsfiles/images/2020/09/202009111821171194_5020_1627156_3.jpg[/img]H5亚型Re-8抗原扩增曲线[img]https://ng1.17img.cn/bbsfiles/images/2020/09/202009111821173647_113_1627156_3.jpg[/img]H5亚型Re-11抗原扩增曲线[img]https://ng1.17img.cn/bbsfiles/images/2020/09/202009111821176118_9242_1627156_3.jpg[/img]H5亚型Re-12抗原扩增曲线[img]https://ng1.17img.cn/bbsfiles/images/2020/09/202009111821177169_9932_1627156_3.jpg[/img]H7N9抗原扩增曲线[img]https://ng1.17img.cn/bbsfiles/images/2020/09/202009111821178224_1520_1627156_3.jpg[/img]H7N9 Re-2抗原扩增曲线5 阳性质控的制备根据GB/T 27539-2011 《动物流感检测 A型流感病毒通用荧光RT-[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]检测方法》规定,阳性对照应Ct28。结合各抗原扩增情况,挑选了H5亚型Re-8、Re-11、Re-12三种抗原适当稀释后进行再次检测,结果如下:[table][tr][td][align=center]稀释倍数[/align][/td][td][align=center]1:10^3[/align][/td][td][align=center]1:10^4[/align][/td][td][align=center]1:10^5[/align][/td][td][align=center]1:10^6[/align][/td][/tr][tr][td][align=center]H5Re-8抗原[/align][/td][td][align=center]16.81[/align][/td][td][align=center]22.18[/align][/td][td][align=center]25.39[/align][/td][td][align=center]28.97[/align][/td][/tr][tr][td][align=center]H5Re-11抗原[/align][/td][td][align=center]15.75[/align][/td][td][align=center]20.79[/align][/td][td][align=center]24.82[/align][/td][td][align=center]28.31[/align][/td][/tr][tr][td][align=center]H5Re-12抗原[/align][/td][td][align=center]14.75[/align][/td][td][align=center]19.72[/align][/td][td][align=center]22.90[/align][/td][td][align=center]26.8[/align][/td][/tr][/table]经综合判定,最后采用H5亚型Re-11抗原作1: 10^5稀释,分装为200μL/管作为阳性质控,Ct值约25,-70℃冻存备用。6 讨论6.1 检测工作更加方便高效一是在核酸提取环节采用了DNA/RNA共提试剂,待检样品经一次提取核酸,除可应用于A型流感等RNA病毒项目检测外,还可应用于非洲猪瘟、圆环病毒等DNA病毒项目检测;二是由于实验室储备了足够量的qRT-[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]预混液及逆转录酶,在开展检测时,不必担心部分检测量较少的项目由于试剂组分不足而罢工事件的发生;三是由于qRT-[url=https://insevent.instrument.com.cn/t/jp][color=#3333ff]PCR[/color][/url]预混液及逆转录酶适用于多数RNA病毒项目的检测,在检测任务发生重大变化时,可以方便地调整检测项目,不必担心试剂不足或浪费情况的发生;6.2 阳性质控品制备方便动物疫病监测实验室常备禽流感血凝抑制抗原,并可根据病毒变异情况及时更新。只需对贮存的抗原进行含量测定,取合适的抗原进行一定比例的稀释即可作为实验室内部的阳性质控品。6.3 适用性有待进一步验证尽管在构建新方法时,对适用性进行了尽可能多的验证,但对于其他毒株,尤其是较新型的变异株是否有效,尚有待进一步观察和验证。6.4 严格依法依规操作如果有关法律法规或上级有关部门出台政策,要求采用获批准的成品试剂盒用于检测,则实验室必须及时停止自建方法的使用。
各位大神,我是第三方检测单位的小白,单位想要扩增资质,想问下电磁辐射、电离辐射这种需要做方法验证吗?如果需要的话该怎么做
PCR技术具有高度的特异性、选择性、灵敏性,而且快速、简便、易于自动化,因此在临床医学工作中受到广泛的重视及合理的应用。目前在我国许多基层医疗单位均已相继开展PCR研究用应用工作。PCR技术很简单,原理也很清楚,因此有条件的单位都能较顺利地上马,但毕竟PCR技术是一种新生事物,受到许多条件因素的影响,所以要做得好也不容易。在PCR技术应用过程中会遇到这样或那样的问题,更甚至会得到与事实完成相反的结果。为了使我们能够顺利地开展PCR工作,更好地发挥PCR技术的工具作用,我们根据多年来PCR工作总结的经验,对PCR扩增过程中经常出现的一些问题进行简要的总结,提供给广大科研工作者作为参考,抛砖引玉,不足之处欢迎指正。 一、 假阳性扩增 在实验过程中有时会碰到所检验的样本全部阳性,而且扩增产物电泳条带亮度均一,这是扩增系统受到污染时最典型的一种表现,需仔细检查,排除污染源,一般应从以下步骤着手: ⒈ 试剂污染: 厂方提供的试剂或其中的某种组分在出厂或运输、贮存过程中受到污染。检测方法,可按试剂盒说明书将试剂分装好,直接置于PCR仪中扩增(不加阴性对照,可加适量蒸馏水),便可简单而有效地判断试剂是否已受到污染。 ⒉ 实验室污染: 这是最常见的污染源,由于PCR技术可以在几个小时内将模板中某些特异性序列扩增到数百万倍以上,扩弗产物可能在电泳等过程中污染[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]、操作台或随着蒸发所形成气溶胶而污染整个实验室。扩增产物又是最有效的模板,一旦出现产物污染其污染程度都较严重。判断方法:可以将实验中最关键步骤如试剂分装、样品处理等转移到一个新的环境中或转移到超净工作台中完成。当然,所用的[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]、吸咀管(离心管)都应彻底更换。解决方法:实验室污染是PCR扩增过程中最易出现的现象,而且一旦出现污染,消除污染源又极其困难,往往需对整个实验室及实验器材彻底清洗处理,所以应该以预防为主,实验过程中严格遵守实验基本要求,样品处理与扩增产物及电泳应尽量分开,最好能在两个房间进行。扩增前处理所用的[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]及扩增后点样用的[url=https://insevent.instrument.com.cn/t/9p][color=#3333ff][url=https://insevent.instrument.com.cn/t/9p][color=#3333ff]移液器[/color][/url][/color][/url]应严格地分开,不可互换。PCR室应保持良好的通风、清洁、最好能专用。 ⒊ 采样污染: 在实验过程中,有时会出现一批结果阳性率很高,一批结果阳性率又下降很多,如此反复,这种现象大都是由于采样时污染所致,一般来讲正规试剂生产厂家,其试剂出厂时都要经过严格的质量检测、批间,特别是批内差异都极小,不致出现如此明显的反复,这种现象主要是由于收集样本的试管或吸管受到污染所致。PCR扩增非常敏感,而一般实验室对重复使用的试管所进行的洗涤方法往注不能去除痕量DNA,这些痕量DNA便成为PCR模板,出现阳性扩增结果,由于采样试管受污染程度不一,所以出现忽高忽低的现象解决方法建议使用一次试管及吸咀取样。 二、 假阴性扩增: 如果连续几次扩增结果都是阴性,或已知阳性样本出现阴性扩增结果,一般认为这是假阴性,出现这现象有以下几种原因: ⒈ 仪器故障: 出现全阴结果,首先考虑到仪器运转是否正常。正确判断仪器的工作状况,是排除假阴性其它原因的基础,仪器运转是否正常是PCR扩增最关键的步聚之一。判断仪器是否正常,首先要测定加热体的温度是否准确,波动范围是否答合要求,各孔之间差异是否符合要求,PCR扩增往往对仪器加热温度及各管间的差异要求有很高的精度,如果误差太大,势必出现假阴性。必须注意的是不能过信名牌,我们经常发现一些名牌机器温度差异高达二度以上。 ⒉ 试剂失效或无效: 了解机器是否正常,便可对试剂进行检测。首先要了解试剂是否超过有效期,试存放方法是否达到要求,如果试剂盒在有效期内,贮存方法是正确,那么就应该仔细、慎重地判断试剂是否是无效试剂或称不合格试剂。可以用已知阳性样本,或试剂盒提供的阳性对照,严格按说明书操作扩增一次,然后把扩增产物用双蒸水稀释1000倍,作为模板进行第二次扩增,判断结果,如果是阴性说明试剂无效或不合格,如果结果阳性那么可用以下方法判定试剂的灵敏度。
一、实验目的 本实验目的了解PCR反应用来扩增DNA 特异性片段实验的的基本原理、实验方法和操作步骤,熟练掌握PCR反应基本体系配制和实验条件的设定。 二、实验原理 PCR(Polymerase Chain Reaction)即聚合酶链式反应是1986 年由Kallis Mullis 发现。这项技术已广泛地应用于分子生物学各个领域,它不仅可用于基因分离克隆和核酸序列分析,还可用于突变体和重组体的构建,基因表达调控的研究,基因多态性的分析,遗传病和传染病诊断,肿瘤机制探查,法医鉴定等方面。PCR技术已成为方法学上的一次革命,它必将大大推动分子生物学各学科的研究发展。PCR是一种利用两种与相反链杂交并附着于靶DNA两侧的寡核苷酸引物经酶促合成特异的DNA 片段的体外方法,由高温变性,低温退火和适温延伸等几步反应组成一个循环,然后反复进行,使目的的DNA 得以迅速扩增,主要过程如图6。置待扩增DNA 于高温下解链成为单链DNA 模板;人工合成的两个寡核苷酸引物在低温条件下分别与目的片段两侧的两条链互补结合;DNA聚合酶在72 ℃将单核苷酸从引物3"端开始掺入,沿模板5"—3"方向延伸,合成DNA 新链。由于每一循环所产生的DNA均能成为下一次循环的模板,所以PCR 产物以指数方式增加,经25—30次周期之后,理论上可增加109倍,实际上可增加107倍。PCR技术具有操作简便、省时、灵敏度高特异性强和对原始材料质量要求低等优点,但由于所用的TaqDNA 聚合酶缺乏5"—3"核酶外切酶活性,不能纠正反应中发生的错误核苷酸掺入,估计每9000个核苷酸会导致一个掺入错误,不过Innis M·A 发现,错误掺入的碱基有终止链延伸的作用倾向,使得错误不会扩大。PCR技术应用广泛,不可能有这样一套条件满足所有的实验,但本实验所介绍的方法可适应于大多数DNA 扩增反应,即使有的不适应,至少也确定了一个共同的起点,在此基础上可以作多种变化。不过下列因素在实验应用时应予以特别注意,以求取得满意结果。1. 模板:单、双链DNA 和RNA都可以作为PCR样品,若起始材料是RNA,须先通过逆转录得取第一条cDNA。虽然PCR 可以仅用极微量的样品,但为了保证反应的特异性,一般宜用ng 量级的克隆DNA,ug 级的染色体DNA,待扩增样品质量要求较低,但不能混合有任何蛋白酶、核酸酶,Taq DNA聚合酶的抑制剂以及能结合DNA 的蛋白质。2. 引物:引物是决定PCR 结果的关键,下列原则有助于引物的合理设计。(1)尽可能选择碱基随机分布,GC 含量类似于被扩增片段的引物,尽量避免具有多聚嘌呤、多聚嘧啶或其它异常序列的引物。(2)避免具有明显二级结构(尤其是在引物3"—末端)的序列。(3)防止引物间的互补,特别要注意避免具有3"末端重叠的序列。(4)引物的长度约为20个碱基,较长引物较好,但成本增加,短引物则特异性降低。(5)引物浓度不宜偏高,过高易形成二聚体,而且扩增微量靶目标或起始材料是粗制品,容易产生非特异产物。3. 缓冲液:PCR缓冲液的变化通常会影响扩增结果,特别是MgCl2,其浓度对专一性和扩增量有重大影响,通常最适浓度为1.5 mM左右(每种dNTP 的浓度为0.2 mM时),浓度过高,使反应特异性降低;浓度过低,使产物产量降低。四种dNTP 浓度通常每种都是0.05 mM—0.2 mM。过高的浓度会导致错误掺入,浓度过低,则影响反应产物的产量。四种dNTP浓度应大体相同,其中一种若偏高,会诱发错误掺入,降低合成速度,过早终止延伸反应。另外dNTP 能与Mg2+结合,使游离Mg2+浓度降低,所以如果dNTP 的浓度有很大改变,MgCl2浓度也要改变。Taq聚合酶是一种耐高温聚合酶,用量通常是1—4 单位/100 ul,浓度过高,产生过多的非特异片段。4. 循环参数:PCR 循环是把起始材料加热到90—95 ℃,保持短时间使双链DNA 解链;然后冷却至37—55 ℃,使引物与模板退火;再升温至70—75 ℃,在TaqDNA聚合酶的作用下掺入单核苷酸,使引物沿模板延伸。解链不完全是导致PCR 失败的最主要原因。用DNA扩增仪时,94 ℃保持1 分钟可使模板的起始物完全变性。若用低于94 ℃的条件,则应适当延长时间。引物与模板退火温度由引物的长度及G+C含量决定。适时间退火(1—2)分钟有利于产物的特异性。引物延伸在70—75 ℃保温的时间可根据扩增DNA片段的长短来调节。正常情况下,每分钟可延伸1 Kb 的长度,常规PCR 一般为25—40个循环,若循环加长,则由于酶活性降低,聚合时间延长,引物及单核苷酸减少等原因,反应后期容易产生错误掺入,所以在满足产物得率前提下,应尽量减少周期次数。三、材料(一)仪器与器皿PRC 扩增仪(PE2400),琼脂糖凝胶电泳设备,微量取样器,一次性指形管,凝胶成像仪 玻片(二)试剂与材料1. 琼脂糖凝胶电泳试剂1)电泳缓冲液:Tris—乙酸0.04 mol/L PH8.0 0.002 mol/L EDTA2)加样缓冲液:0.25%溴酚兰40% w/v蔗糖3)溴化乙锭溶液:0.05 mg/ml溴化乙锭/水4)琼脂糖2. TaqDNA 多聚酶3. 5′反应缓冲液:125 mmol/L Tris-HCl pH8.2;10 mmol/L MgCl2;0.5 mg/ml gelatin;125 mmol/L(NH4)2SO4; Formamide 25%4. 混合dNTP 液(dATP dGTP dTTP dCTP各2 mmol/L)5. DNA 模板(每2 ml 中含有10 fg 待扩增DNA)6. 引物 1 (25 pmol/L),5’加入EcoRI 粘性末端碱基7. 引物 2 (25 pmol/L),5’加入HindIII 粘性末端碱基8. 无菌水四、实验步骤1. 按顺序在200 ml 指形管中加入以下试剂与样品:(因购入的试剂批次不同,加样时有 所差别,以预实验结果为准。)1) ddH2O 74 ml2) 10′Buffer 10 ml3) MgCl2 6 ml(10′Buffer 如已加入MgCl2,则不必加)4) dNTP 2 ml5) 引物1 2 ml6) 引物2 2 ml7) 模板 2 ml8) Taq DNA聚合酶2 ml总体积共100 ml(也可以配成40 ml 的反应体系)2. 在PCR 扩增仪上按以下反应条件编入程序:(以下为参考值,因扩增的DNA片段不同,各类PCR 扩增仪程序设定各不相同,编程过程视扩增的DNA 片段的要求及仪器而定参数,见示范。)预变性 94 ℃ 2 分种循环条件(30 次)变性 94 ℃ 40 秒复性 55 ℃ 35 秒延伸 72 ℃ 2 分10 秒延长延伸 72 ℃ 7 分钟编完反应程序,置反应管于PCR扩增仪的反应孔中,开动机器,扩增循环反应开始。3. PCR 扩增完毕,配2%琼脂糖凝胶,取15 ml 反应液及相适应的PCR mark 分别点样, 加样缓冲液应为40%W/V 蔗糖,电泳观察结果。4. 凝胶成像仪或紫外灯下观察实验结果,是否已扩增到实验设计的DNA 片段。注意事项: 要想得到预期的实验结果,PCR的反应条件和很多参数有着密切的关系,都应引起注意。在实验设计中我们要考虑到模板浓度的大小,设计引物时也要主要引物长度适当以及恰当的GC含量,在PCR体系中引物浓度,dNTPs浓度相对要均衡,选取的taq enzyme,缓冲液的离子含量也是影响产物的结果,另外就是要根据产物大小设定适当的变性时间,退火温度及时间,延伸的时间。
各位大神,我是第三方检测单位的小白,单位想要扩增资质,想问下电磁辐射、电离辐射这种需要做方法验证吗?如果需要的话该怎么做
仪器简介PCR是利用DNA聚合酶对特定基因做体外或试管内In Vitro的大量合成,它是利用DNA聚合酶进行专一性的连锁复制。目前常用的技术,可以将一段基因复制为原来的一百亿至一千亿倍。根据DNA扩增的目的和检测的标准,可以将PCR仪分为普通PCR仪,梯度PCR仪,原位PCR仪,实时荧光定量PCR仪四类。PCR技术原理该技术是在模板DNA、引物和四种脱氧核糖核苷酸存在下,依赖于DNA聚合酶的酶促合成反应。DNA聚合酶以单链DNA为模板,借助一小段双链DNA来启动合成,通过一个或两个人工合成的寡核苷酸引物与单链DNA模板中的一段互补序列结合,形成部分双链。在适宜的温度和环境下,DNA聚合酶将脱氧单核苷酸加到引物3´-OH末端,并以此为起始点,沿模板5´→3´方向延伸,合成一条新的DNA互补链。◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆◆列举部分仪器的个别参数,仅供参考:技术参数:样品台容量:96X0.2ml PCR管、 96孔PCR板、8排/12排PCR管工作原理:TE半导体系加热致冷技术屏幕:彩色触摸屏(5.7英寸)温度控制范围:4℃-105℃ 升降温速度:≥4.0℃/秒 温度均匀性:≤0.3℃ 温控精度:≤0.1℃ (@55℃) / ≤0.2℃(@90℃以上)温度显示精度:0.1℃程序温控方式:Block模式和Tube模式变温速度可调范围:0.1-4.0℃程序可储存数:最大可存250个程序,可通过U盘扩展存贮最大循环数:99循环,可套嵌两级,可做巢式PCR实验时间递增/递减:0-9分59秒,可做Long PCR实验温度递增/递减:0.1-9.9℃,可做Touch down实验断电保护/暂停功能:有Soak功能:有运行状态显示功能:[color=







 我要推广仪器
我要推广仪器
 下载APP
下载APP