现在需要进行一个液相色谱柱的封端的培训,有哪位高手知道不同填料的色谱柱采用何种封端,或者有相关的资料的可以提供一下吗?
在液相上用氨基柱做维生素E的检测,四个目标峰和溶剂峰都出现了双峰问题,是不是柱头塌陷造成的,该怎么处理呢?
[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=16722]关于液相色谱柱的原理、键合、封端、评价标准的ppt文件,希望对大家有用!![/url]关于液相色谱柱的原理、键合、封端、评价标准的ppt文件,希望对大家有用!!
液相色谱峰形拖峰与预柱的判断及处理一、案例介绍:l 测定油悬浮剂中的有效成份:双草醚;l 采用国产仪器伍丰LC-100分别搭配250×4.6mmODS柱和250×4.6mm BRISA LC2 C18柱,对应流动相比例分别为甲:乙:水=20:35:45和甲:乙:水=20:40:40,检测波长为246nm;l 发生该异常(严重的峰形拖尾)状况时,仪器可见部分均正常运行,系统反馈压力较正常的上升2.0~3.2Mpa,其它操作均符合实验操作之要求;l 按标准操作所得液相图谱峰形异常,主要是拖尾因子1.00变化至1.77;二、案例图谱标准图谱:http://ng1.17img.cn/bbsfiles/images/2017/01/201701191700_668316_2239775_3.png双草醚-液相分析标准图谱 1拖尾因子1.00注:实验条件250×4.6mm BRISA LC2 C18柱,流动相甲:乙:水=20:40:40,检测波长为246nm,目标峰保留时间为16.232min异常图谱:(不可接受)http://ng1.17img.cn/bbsfiles/images/2017/10/2016071409341955_01_2239775_3.png双草醚-液相异常分析图谱1 拖尾因子1.77 注:实验条件250×4.6mm BRISA LC2 C18柱,流动相甲:乙:水=20:40:40,检测波长为246nm,目标峰保留时间为16.232minhttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071409345934_01_2239775_3.png双草醚-液相异常分析图谱2 拖尾因子1.79 注:实验条件250×4.6mm BRISA LC2 C18柱,流动相甲:乙:水=20:35:45,检测波长为246nm,目标峰保留时间为19.240minhttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071409353819_01_2239775_3.png双草醚-液相异常分析图谱3-1 拖尾因子1.28注:实验条件250×4.6mm ODS柱,流动相甲:乙:水=20:35:45,检测波长为246nm,目标峰保留时间为16.407minhttp://ng1.17img.cn/bbsfiles/images/2017/10/2016071409361418_01_2239775_3.png双草醚-液相异常分析图谱3-2 拖尾因子1.28(上图目标峰起始部分和结束部分放大图)三、异常问题与分析1. 异常问题简述: 在进行正确的操作后,通过观察双草醚液相分析的图谱发现在同条件下所得目标峰的保留时间无明显变化,但出现明显或不可接受的峰形拖尾现象。按经验调整流动相中有机相(降低5%)与水相(增加5%)的比例再次进样分析,所得图谱发现目标峰的保留时间有明显变化但峰形的拖尾因子仍未得到改善。即正常情况下和微调后无法对分析条件进行优化。2. 原因分析:http://ng1.17img.cn/bbsfiles/images/2017/10/2016071409371743_01_2239775_3.png 鉴于液相分析条件的确定性(科学合理)、人为操作的正确性(准确无误)、色谱柱的使用寿命(才启用8个月)以系统运行压力的异常结果(正常压力为16.3~16.6Mpa上升到18.3Mpa及以上),故判断为流动相传输系统异常故障。四、异常状况处理http://ng1.17img.cn/bbsfiles/images/2016/07/201607140938_600425_2984502_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607140938_600426_2984502_3.png更换新的预柱1注:更换预柱柱芯后之所以更换整个预柱主要是为止先前的柱套内仍有异物,如此可避免产生污染,从而达到更换预柱柱芯的目的;旧的预柱可超声清洗后留待备用……装上新预柱后,顺道把色谱柱反冲洗了一下……然后……然后就好……五、异常状况处理之结果 经处理后液相分析所得图谱恢复正常(0.95≤拖尾因子≤1.05),确定是预柱内有异物造成流动相传输系统故障导致系统压力升高和目标峰峰形严重拖尾。不可以打脸……
做高效液相的时候柱压开始的时候不稳定,连续上下跳动,流动相走很长一段时间(半小时)才趋于稳定,不知道啥原因。几天前做的样品,今天做同样的样品,柱压就不一样了,出峰时间也就不一样了,(流动相条件一样,检测波长一样……),虽然可以辨别主峰是哪个,但是往文章上面贴图,恐怕不好。请问,这个问题是怎么回事?液相的问题还是柱子的问题?流动相是甲醇和水,过膜、脱气处理过,流动相应该没有问题。
液相色谱柱才用了两个月,出峰拖尾是什么情况?如何解决?
液相色谱柱,主峰拖尾比较严重了怎么处理,如果在不购买柱子的情况下 C18柱4.6*250mm*5um
[color=#444444]安捷伦1220液相色谱柱压一直在110~130之间起伏(偏高),是什么原因导致的?[/color][color=#444444]峰形出现过正常峰,但保留时间与正常值有偏差,后来又打了几个样,发现直接不出现峰了,这是出现什么问题,都有哪几方面的可能?解决方法?[/color]
液相柱后衍生做氨基甲酸酯,溶剂峰一直前移,溶剂峰是倒峰,没法正常做样,求各位大神指点一下
如题,液相色谱中,主成分在该杂质的最大吸收波长下是有吸收的,该杂质在此条件下出峰都正常。改换流动相之后,主成分出峰。如果在原条件下进行分析会对该杂质的检测结果有何影响吗?主成分一定要出峰吗?
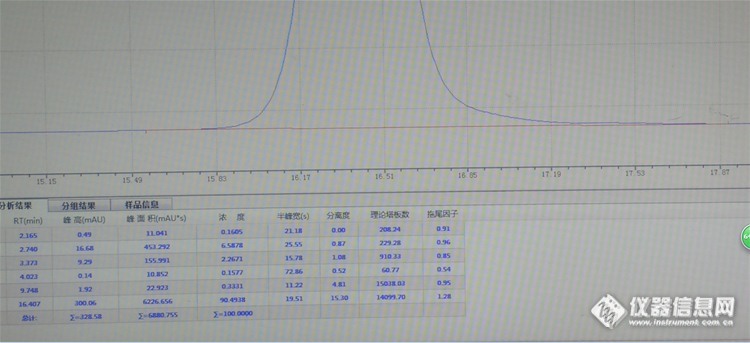
求助,我用液相正常色谱条件下走的对照品,前20分钟只有几个峰,后面冲柱子的几分钟出现了好多峰,不知道是哪里出了问题……详情见图[img=,690,299]https://ng1.17img.cn/bbsfiles/images/2019/07/201907140822138097_8039_3369278_3.jpg!w690x299.jpg[/img][img=,690,440]https://ng1.17img.cn/bbsfiles/images/2019/07/201907140822175530_7381_3369278_3.jpg!w690x440.jpg[/img]
岛津10A的液丰,2台。同一样品,相同流动相,相同时间进样,一台液相峰面积一直稳定,而一台峰面积变化一直很大,多次实验都是这样,两台更换色谱柱,峰面积稳定的还是稳定,不稳定的还是不稳定。 换用另一台岛津的液相和PE白液相峰面积仍不稳定。请问高手是是什么原因
液相色谱出峰顺序和反相液相色谱出峰顺序有何不同同一组物质,采用正相液相色谱和反相高效液相色谱柱分别进行分离,出峰的顺序是不是会相反啊,比如使用正相色谱柱时是1、2、3、4、5种物质的出峰顺序为12345,而用反相柱时出峰顺序会变成54321啊,谢谢!
我们有一台岛津10A的液相色谱,平时使用的都很正常,最近总是出现峰型拖尾的现象,排除柱子与样品的问题,检测器的流通池也洗过了,还有检测器的能量有800,基线正常,不知道是什么原因
[color=#444444]安捷伦高效液相 [/color][color=#444444]色谱条件:流动相:甲醇:0.2%磷酸水(40: 60);流速:1 mL/min; 检测波长:225 nm;柱温:35 ℃;进样体积:20 μL[/color][color=#444444]以前相同条件下做的时候柱压一般会在190~220 bar之间,现在做了几次最初达到平衡的柱压都只有170 bar左右,更奇怪的是进完样品后柱压就开始下降,基线下移,2~3 min会出一个溶剂峰,再后面柱压降到120 bar左右不变,基线下移厉害,在负值处走平,样品峰一直不出现,这大概会是什么原因呢?[/color]
高效液相为什么柱子加长峰变窄保留时间不变
高效液相柱后衍生测氨基甲酸酯峰太宽。柱后衍生试剂用皮克林原装进口的,色谱柱用氨基甲酸酯分析专用柱3.9*150,进样量10ul,脱洗梯度在761的基础上优化了一下,峰太宽了,每个完整的峰都要1min多时间,而且还有拖尾现象,滴灭威亚砜和滴灭威砜完全分不开,求各位大神指导,妹纸在这感恩不尽
这几天在做液相制备,发现一个怪现象,柱子平衡好之后,每次进样,峰的重复性都比较好,都能对应的找到上一针的峰,但是,每个峰的面积都逐渐变小了,比如说第一针的时候有33,第二针的时候只有17,第三针的时候只有10,而且是所有的峰都一针一针的逐渐变小了,而进样量每次都是一样的,请教各位,这是怎么回事呢?
同一色谱柱下和同一液相条件下近期色谱峰峰宽变宽?为什么?柱子用久了?
[b][color=#444444] [/color][color=#444444]之前我们的色谱柱有拆下来过,昨天接上色谱柱,重新配置新的流动相跑了一下午基线,基线都比较平稳。但是今天用就一直状况百出,早上用的时候仪器启动后缓启动一分钟液相柱进口漏液,蒸发光检测器压力没有显示。后面调了空气压缩泵的压力蒸发光检测器压力有显示,液相柱重新拧紧后不漏液,走基线还算平稳。进了一个8毫克每毫升的样品,平时十多分钟就能出峰跑了半小时不见出峰。重新跑基线,基线呈锯齿形浮动,基线不平稳,[/color][color=#444444] [/color][color=#444444]发现液相柱出口漏液, 拧紧重新走基线还算平稳,下午接连几个样都不出峰。我们进的样是用标准品配的,所以浓度应 该不算低,检测器的数据也在正常范围内,恒流泵上的压力在7-9左右,也算正常,有哪些原因导致液相不出峰呢[/color][/b]
新的C18液相色谱柱空白出峰是什么原因
请问各位大神,我现在遇到这种情况,用液相走样品,样品沸点大概有600,C18柱,纯乙腈流动相,但出峰时间在40min左右,调大流速,其杂质峰不出现,请问这种情况下怎么解决!
我们本来用的是waters液相自动进样,现在换成岛津手动进样,用的是同一根柱子,流动相比例也相同,但是有一个物质出峰事件相差很大(本来waters仪器出峰大概9min现在岛津仪器出峰在2.1min),其余物质出峰时间相差不大,各位大神知道这是什么原因吗?
我们使用的日立液相,走空白走不好,太多杂峰,但是同样的柱子和条件在别的液相走的很好 怎么处理
液相色谱,走空白走不好,太多杂峰,但是同样的柱子和条件在别的液相走的很好,是什么原因呢?
这几天在做液相制备,发现一个怪现象,柱子平衡好之后,每次进样,峰的重复性都比较好,都能对应的找到上一针的峰,但是,每个峰的面积都逐渐变小了,比如说第一针的时候有33,第二针的时候只有17,第三针的时候只有10,而且是所有的峰都一针一针的逐渐变小了,而进样量每次都是一样的,请教各位,这是怎么回事呢?
同一批样品之前用Water液相,C18柱子(旧的)分析,有3个峰,依次称为1号,2号,3号峰,3个峰之间保留时间间隔1-2分钟,现在同一个样换了一台Water液相和同品牌同型号的新C18柱子分析时2号,3号峰的位置和峰面积基本没变,1号峰的峰面积变小好多而且在3号峰后面出了一个大的峰,3个峰保留时间的间隔还是1-2分钟,请问有人碰到过这样的情况吗?求帮助!
液相色谱仪由高压液体泵、检测器及液相色谱柱等三部分组成,其中液相色谱柱的正确安装和使用,是液相色谱工作的关键;也是液相色谱工作者获得正确可靠的实验数据的必经之路。 一、液相色谱柱的安装: 1、液相色谱柱的结构: a、空柱由柱接头、柱管及滤片组装而成。 柱接头采用低死体积结构,柱接头是两端螺纹组件,一端是为7/16英寸外螺纹,另一端是3/16英寸的内螺纹(国内外已规范化)。7/16英寸外螺纹与1/4英寸柱管(Φ6.35mm)连接,中间放置压坏用于密封。3/16英寸的内螺纹与1/16英寸(Φ1.57mm)的连接管连接,中间也放置压环用于柱接头的密封。为了尽量减少柱外死体积,在安装色谱柱时,用Φ1.57mm连接管通过空心螺钉压环后要尽量插到底,然后再拧紧空心螺钉。压环被空心螺钉挤压变形后紧箍在连接管上(连接管通过压环后露出的管长度应严格控制在2.5mm长或其他固定尺寸)。 在两端柱接头内,柱管两端各放置一片不锈钢滤片(或滤网),用于封堵柱填料不被流动相冲出柱外而流失。空柱各组件均为316#不锈钢材质,能耐受一般的溶剂作用。但由于含氯化物的溶剂对其有一定的腐蚀性,故使用时要注意,柱及连接管内不能长时间存留此类溶剂,以避免腐蚀。 b、柱填料: 液相色谱柱的分离作用是在填料与流动相之间进行的,柱子的分类是依据填料类型而定。 正相柱:多以硅胶为柱填料。根据外型可分为无定型和球型两种,其颗粒直径在3—10 µm的范围内。另一类正相填料是硅胶表面键合—CN,-NH2等官能团即所谓的键合相硅胶。 反相柱:主要是以硅胶为基质,在其表面键合十八烷基官能团(ODS)的非极性填料。也有无定型和球型之分。 常用的其他的反相填料还有键合C8、C4、C2、苯基等,其颗粒粒径在3—10 µm之间。 2[/font

我们公司安捷伦的1200液相。最近出了一个比较怪的现象。本司为第三方,最近有一个比较怪的情况。同种类型的柱子,长柱子可以出峰,短柱子不能出峰,而且我们特意新买的一根全新的短柱子,走了还是不能出峰。之前我们无论是长柱还是短柱都是可以出峰的。柱子型号和参数:长柱:XDB-C18 5um,4.6*250mm,另外一个是短柱XDB-C18 5um,4.6*150mm,也就是一根是250mm的柱子,一个是150mm的柱子。现在这种情况不知道为什么什么。求助各位。我们已经排除了流动相、泵等其它方面的影响。色谱 图如下:http://ng1.17img.cn/bbsfiles/images/2017/10/2015080317193350_01_1786331_3.jpgfile:///C:\Documents and Settings\Administrator\Application Data\Tencent\Users\234809767\QQ\WinTemp\RichOle\CTQG))90XG((]OW(T)YBTPP.jpgfile:///C:\Documents and Settings\Administrator\Application Data\Tencent\Users\234809767\QQ\WinTemp\RichOle\CTQG))90XG((]OW(T)YBTPP.jpg 正常的峰[img=,455,602]http://ng1.17img.cn/bbsfiles/images/2015/08/201508031722_558891_1786331_3.png 完全不出峰
我用液相PDA检测器做邻苯二甲酸酯,流动相甲醇-水。浓度低的时候在目标峰位置出现倒峰,改变色谱条件仍然跟标样在相同保留时间处有倒峰,并且倒峰的最大吸收波长也与标样一样。进甲醇,乙腈也有倒峰,进水是正峰,进空针没有。换过流动相,色谱柱都没有解决。请教大神们,到底怎么回事







 我要推广仪器
我要推广仪器
 下载APP
下载APP