发射光谱半定量分析有哪些具体方法?
光谱 分析有半定量分析吗?
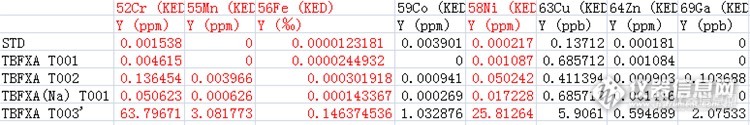
坛里的高僧,给分析下,为什么半定量分析结果与定量分析结果差别会这么大???说好的在30%内。备注:STD---1ppb调谐液(含锂、钴、铟、铀)1、2、4批取0.1g消解后稀释至100ml检测,第3批料液为水溶液,直接进样了。http://ng1.17img.cn/bbsfiles/images/2016/08/201608032051_603285_3124053_3.pnghttp://ng1.17img.cn/bbsfiles/images/2016/08/201608032051_603286_3124053_3.png
ICP光谱仪的应用中有半定量分析一项,看完解释之后,仍然觉得不是很清楚,希望哪位高人能够解释一下,最好能够举一个具体的列子!说明一下什么时候可以采取半定量分析。万分感激!
请教各位专家,如何进行半定量分析?
从原理上讲,原子发射光谱可以进行定性分析、半定量分析和定量分析。过去因为电弧光源和火花光源在性能上存在缺陷,使发射光谱在定性和半定量分析方面应用非常广泛,但其定量分析受到很大限制。ICP光源的出现彻底改变了这种状况,使发射光谱定量分析的应用越来越多,现已基本上成为元素分析的常规手段。但大家在用ICP进行定量分析的同时,还经常去做定性和半定量分析吗?
群里有没有大神对半定量分析比较熟悉的?它存在的理论基础是什么,实际分析中我们如何使用此方法来定量呢?其定量的结果是否只能用作参考?目前有碰到过一硅氧烷的测试,结果中有这么一句话“D8,D9,D10以D5来进行半定量”,很疑惑这种定量方式,盼群里大神们帮忙解惑啊
请问半定量分析用到的标液用多大浓度比较好呢?
都说能谱仪能半定量分析元素,能否比较准确的定量分析呢?下面是我们遇到的问题:一个未知样品,你全面扫描和部分扫描所分析出来的质量百分比是完全不一样的。
看到过许多地方介绍核磁共振用于一般有机化合物的定量分析。但仪分实验课上作出的谱图(乙酰乙酸乙酯的烯醇异构体分析)很不理想,应该成比例的峰面积差很多。请问要想做好定量分析的话需要注意哪些问题呢?顺便问一下,核磁作定量分析的准确度大概能到多少呢?
看到一篇文章说可以用质谱的特征离子半定量分析肉中的香气成分。他也选了一种内标物,再用质谱的几种特征离子就可以进行半定量。但是看的还不是很明白,请教各位有没有做过类似实验的兄弟姐妹,还望指点一二。谢谢!
红外能做半定量分析吗?在我的软件上没有看到怎么计算吸收峰的峰面积.你们的软件上有如何计算红外吸收峰的峰面积吗?
红外能做半定量分析吗?在我的软件上没有看到怎么计算吸收峰的峰面积.你们的软件上有如何计算红外吸收峰的峰面积吗?
各位同行,本人用的是JY公司的ULTIMA2型ICP,以前只是做定量分析,现在想研究一下如何进行半定量分析,请问哪位有没有这方面的资料,谢谢了.本人在广州市,有没有同在这儿的朋友呢?
红外光谱的定量分析简述[img]http://www.instrument.com.cn/bbs/images/affix.gif[/img][url=http://www.instrument.com.cn/bbs/download.asp?ID=19088]红外光谱的定量分析[/url]
大家好,请问一下大家能介绍一下光谱分析的定量依据吗?我了解的光谱就是发射光谱和吸收光谱,吸收光谱就是[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url],发射光谱就是ICP,我觉得原子荧光也算发射吧?不知道原子荧光算那种还是新的名称? 我的理解是,吸收光谱分析的依据应该是朗伯-比尔定率,因为它就是一个入射光通过一个均匀介质(一定厚度的原子密度),利用吸收的光与[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]成正比关系,符合朗伯-比尔定率。 那发射光谱的定量依据是什么?最近在网上看到说的是罗马金-赛伯公式,具体是:光谱定量分析依据试样中欲测元素的谱线强度来确定元素的浓度。元素的谱线强度I与该元素在试样中的浓度c的关系为:I=ACb,这个公式称为罗马金-赛伯公式,是光谱定量分析的基本公式。式中A及b是两个常数。常数A是与试样的蒸发、激发过程和试样组成等有关的一个参数;常数b称为自吸系数,它的数值与谱线的自吸收有关。所以只有控制在一定的条件下,在一定的待测元素含量的范围内,A和b才是常数。取对数得光谱定量分析的基本关系式lgI=blgC+lgA。 不知道我的理解是不是正确?不知道大家有什么想法或补充的,大家讨论讨论吧!!谢谢
光谱怎么进行定量分析?
谁能提供一些有关质谱定量分析的经验?谢谢!!
请教大家,如何对一生物组织体的荧光光谱进行定量分析呢,通常,我们说的定量分析是对荧光组分的定量分析,那是如何实现的,请教,我的e-mail:lls009663@163.com
近红外光谱定量分析中,定量模型的评价有两个指标RMSEP、RMSEC,为什么一般RMSEP的值大于RMSEC
我是一名医学在校研究生,现在想做一红外光谱分析仪测尿路结石成分定量分析的实验。我们研究所里没有定量分析的软件,请问各位前辈,对于你们专业人士,定量分析难吗?有哪位愿意帮助我分析的呢?我可以付酬金。
瓦里安710的 ICP半定量分析在哪设置
需要定量分析CH4,CO2,CO,H2,反应是CH4+CO2=2CO+2H2,正在想法对其进行标定,得出反应的转化率。使用CH4,CO2,CO,H2纯气,按比例混合后进入质谱进行分析,如何进行试验数据处理,如何进行试验能使标定误差最小?求高人指点
无标半定量分析可以直接选定几个元素,建立方法,然后定性分析,得出结果可以用全扫描,把所有的元素选出,建立方法,定性分析,也会得到无标半定量的结果理论上,这两个数值,应该没有大的差异可是实际上,却相反想请教一下大家
关于液相色谱的定量分析定量分析是在定性分析的基础上,需要纯物质作为标准样品。液相色谱的定量是相对的定量方法,即:由已知的标准样品推算出被测样品的量。
内标法与外标法一、内标法 什么叫内标法?怎样选择内标物? 内标法是一种间接或相对的校准方法。在分析测定样品中某组分含量时,加入一种内标物质以校谁和消除出于操作条件的波动而对分析结果产生的影响,以提高分析结果的准确度。 内标法在气相色谱定量分析中是一种重要的技术。使用内标法时,在样品中加入一定量的标准物质,它可被色谱拄所分离,又不受试样中其它组分峰的干扰,只要测定内标物和待测组分的峰面积与相对响应值,即可求出待测组分在样品中的百分含量。采用内标法定量时,内标物的选择是一项十分重要的工作。理想地说,内标物应当是一个能得到纯样的己知化合物,这样它能以准确、已知的量加到样品中去,它应当和被分析的样品组分有基本相同或尽可能一致的物理化学性质(如化学结构、极性、挥发度及在溶剂中的溶解度等)、色谱行为和响应特征,最好是被分析物质的一个同系物。当然,在色谱分析条什下,内标物必须能与样品中各组分充分分离。需要指出的是,在少数情况下,分析人员可能比较关心化台物在一个复杂过程中所得到的回收率,此时,他可以使用一种在这种过程中很容易被完全回收的化台物作内标,来测定感兴趣化合物的百分回收率,而不必遵循以上所说的选择原则。 在使用内标法定量时,有哪些因素会影响内标和被测组分的峰高或峰面积的比值? 影响内标和被测组分峰高或峰面积比值的因素主要有化学方面的、色谱方面的和仪器方面的三类。 由化学方面的原因产生的面积比的变化常常在分析重复样品时出现。 化学方面的因素包括: 1、内标物在样品里混合不好; 2、内标物和样品组分之间发生反应, 3、内标物纯度可变等。 对于一个比较成熟的方法来说,色谱方面的问题发生的可能性更大一些,色谱上常见的一些问题(如渗漏)对绝对面积的影响比较大,对面积比的影响则要小一些,但如果绝对面积的变化已大到足以使面积比发生显著变化的程度,那么一定有某个重要的色谱问题存在,比如进样量改变太大,样品组分浓度和内标浓度之间有很大的差别,检测器非线性等。进样量应足够小并保持不变,这样才不致于造成检测器和积分装置饱和。如果认为方法比较可靠,而色谱固看来也是正常的话,应着重检查积分装置和设置、斜率和峰宽定位。对积分装置发生怀疑的最有力的证据是:面积比可变,而峰高比保持相对恒定, 在制作内标标准曲线时应注意什么? 在用内标法做色话定量分析时,先配制一定重量比的被测组分和内标样品的混合物做色谱分析,测量峰面积,做重量比和面积比的关系曲线,此曲线即为标准曲线。在实际样品分析时所采用的色谱条件应尽可能与制作标准曲线时所用的条件一致,因此,在制作标准曲线时,不仅要注明色谱条件(如固定相、柱温、载气流速等),还应注明进样体积和内标物浓度。在制作内标标准曲线时,各点并不完全落在直线上,此时应求出面积比和重量比的比值与其平均位的标准偏差,在使用过程中应定期进行单点校正,若所得值与平均值的偏差小于2,曲线仍可使用,若大于2,则应重作曲线,如果曲线在铰短时期内即产生变动,则不宜使用内标法定量。 二、外标法 用待测组分的纯品作对照物质,以对照物质和样品中待测组分的响应信号相比较进行定量的方法称为外标法。此法可分为工作曲线法及外标一点法等。工作曲线法是用对照物质配制一系列浓度的对照品溶液确定工作曲线,求出斜率、截距。在完全相同的条件下,准确进样与对照品溶液相同体积的样品溶液,根据待测组分的信号,从标准曲线上查出其浓度,或用回归方程计算,工作曲线法也可以用外标二点法代替。通常截距应为零,若不等于零说明存在系统误差。工作曲线的截距为零时,可用外标一点法(直接比较法)定量。 外标一点法是用一种浓度的对照品溶液对比测定样品溶液中i组分的含量。将对照品溶液与样品溶液在相同条件下多次进样,测得峰面积的平均值,用下式计算样品中i组分的量: W=A(W)/(A) 式中W与A分别代表在样品溶液进样体积中所含i组分的重量及相应的峰面积。(W)及(A)分别代表在对照品溶液进样体积中含纯品i组分的重量及相应峰面积。外标法方法简便,不需用校正因子,不论样品中其他组分是否出峰,均可对待测组分定量。但此法的准确性受进样重复性和实验条件稳定性的影响。此外,为了降低外标一点法的实验误差,应尽量使配制的对照品溶液的浓度与样品中组分的浓度相近。 外标法 external standard method 色谱分析中的一种定量方法,它不是把标准物质加入到被测样品中,而是在与被测样品相同的色谱条件下单独测定,把得到的色谱峰面积与被测组分的色谱峰面积进行比较求得被测组分的含量。外标物与被测组分同为一种物质但要求它有一定的纯度,分析时外标物的浓度应与被测物浓度相接近,以利于定量分析的准确性。 三、定量分析中怎样选择内标法或外标法(来源:药物分析网) 选一与欲测组分相近但能完全分离的组分做内标物(当然是样品中没有的组分),然后配制欲测组分和内标物的混合标准溶液,进样得相对校正因子。再将内标物加入欲测组分的样品中,进样后测得欲测组分和内标物的定量参数。用内标法公式计算即可。 内标法是将一定量的纯物质作内标物,加入到准确称量的试样中,根据被测试样和内标物的质量比及其相应的色谱峰面积之比,来计算被测组分的含量。 选择内标物有4个要求:1.内标物应是该试样中不存在的纯物质;2.它必须完全溶于试样中,并与试样中各组分的色谱峰能完全分离;3.加入内标物的量应接近于被测组分;4.色谱峰的位置应与被测组分的色谱峰的位置相近,或在几个被测组分色谱峰中间。 内标法的优点是测定的结果较为准确,由于通过测量内标物及被测组分的峰面积的相对值来进行计算的,因而在一定程度上消除了操作条件等的变化所引起的误差。内标法的缺点是操作程序较为麻烦,每次分析时内标物和试样都要准确称量,有时寻找合适的内标物也有困难。外标法简便,但进样量要求十分准确,要严格控制在与标准物相同的操作条件下进行,否则造成分析误差,得不到准确的测量结果。 内标与外标都是定量的一种方法而已,至于哪一种方法好与不好不能一概而论,做不同的分析,面对着不同的要求,再加上分析成本分析效率等等问题,我想简单而有效进行定量分析来满足要求才是最重要的。1、以前做过很多医药、农药中间体的芳香族卤代化合物的常量定量分析,没有自动进样器,用外标法定量,确实重现性与稳定性非常差,结果经常受到搞合成同事的质疑。其实,仔细分析原因不一定就是外标法不适合这种定量分析,首先我们的实验室仪器和手段是否调整到一种稳定而合理的状态了,比如,衬管是否洁净,玻璃棉的位置是否合适恰当(能否使样品尽可能的汽化)、汽化温度是否合适、色谱峰形是否对称(也就是样品与色谱柱健合相是否匹配)、附近有没有其它色谱峰的干扰、选用什么进样方式(如快速进样还是热针进样)等等因素的影响都需要考虑,如果这些因素都考虑了,按照GMP方法验证对于精密度的要求,同一样品进6针以上的RSD和配制6个样品的定量结果RSD都能满足小于1.5%的要求,那么这个方法用外标法就是完全适用的,但是前面的影响因素是一定要都考虑到的,否则谈论这个方法是否适用就有失偏颇了。在做过的许多出口产品的定量分析方法当中有许多是一些医药公司提供的比较完善而验证过的方法,内标与外标都有(他们用的都是自动进样)精密度都能满足RSD小于1.5%的要求,当一个方法能够满足测试要求的时候,无论内标外标,都是可行的,当然有一个分析成本和分析时间的问题,内标的成本和控制溶液、样品溶液的配制当然要比外标要高和麻烦一些了。而有些时候,可能受你实验室现有仪器和附属设备的影响,达不到一定的要求,而还必须进行定量分析,有时外标的结果可能就要差一些,这时,你可能就要考虑用内标法了,可以排除手动进样的误差、分流歧视的影响、包括一些未知因素平行误差的影响,这时内标可能就显示出它的优势来了。 2、上面已经提到当做方法验证的时候,当同一样品配制6个样品溶液用所选用的外标法进行定量的时候,RSD都满足1.5%的要求时,也分为两种情况,小于1%和大于1%小于1.5%。如果RSD的结果小于1%,那这个方法就没有什么可以怀疑的了;如果RSD的结果大于1%而在1.5%略低一些的范围活动时,这个方法的可行性就将受到质疑,毕竟这是方法验证,你就要考虑上面1所提到的影响因素的影响了,如果排除掉以上的影响因素,RSD还是在1.5%附
我想请教各位大侠有关色谱定量分析的问题!我在做有机硅方面的科研。我不知道需不需要用内标物进行定量?我用的面积归一法。另外,根据色谱图能进行产物的收率和反应物的转化率计算吗?谢谢!
有使用英国hiden公司的hpr20做定量分析的吗?定量分析如何标定?请教
用x’pert highscore 进行物相定量、半定量分析作者wustliang X’pert Highscore 是荷兰philips分析仪器公司推出的一款用于xrd物相分析的软件,简便易学。笔者使用该软件已经有4、5年的时间了,就该软件的使用有一点体会。最近有朋友垂询如何用highscore对物相进行半定量、定量分析,现说一点经验,仅供参考。半定量分析(semi-quantitative analysis) 半定量方法是一种介于定量和定性之间的一种科学方法,主要应用于对某一物理量概念和趋势的快速判定,并通过对总体的分析,快速的求解问题。必须对其特点给予说明:对某些分析准确度要求不高,只能给出其大致含量。在x射线衍射里经常使用RIR参考强度比法。 RIR即参考强度比,很多pdf卡片中都给出了这个值。如21-1152卡片标定的是Al2MgO4(spinel),其RIR为2.13;10-0173的卡片标定的是α-Al2O3(corundum),其RIR为1.00。 RIR的物理意义是该卡片所标定的本相的最强峰和α-Al2O3(corundum)的最强峰的强度比值。如21-1152中的2.13即Al2MgO4的最强峰(311)和α-Al2O3(corundum)的最强峰(113)的强度比。由于种种原因同一种相可能有很多张卡片,而这些卡片所给出的RIR值不尽相同。如quartz有上百张卡片,其中81-1665给出的RIR值为5.06,而79-1901为3.07,75-0443为1.31,75-0335给出的值最小为0.14,相去悬殊。那么我们如何选择呢? 1.首选NBS(美国国家标准)认可的卡片。它的可信程度是很高的,很多文章都引用。 2.其次选择质量标识为S的卡片,它可信程度也很高,也经常被引用。 3.再次选择卡片号比较大的,如80或90开头的卡片。这些卡片制做的年代比较晚,可信程度相对较高。 4.最后还可以对多张卡片的RIR值进行综合的考虑。比如求个平均值,看哪一张卡片的RIR最接近平均值就用哪一张。但一般不要用最高或最低的RIR卡片参与求平均值。 5.特别的是α-Al2O3的RIR值一定要选1。 如果你有一系列的试样分别为A1,A2,A3……An,里面都含有α1、α2和α3相;而且你选择的卡片为: α1:11-xxxx α2:22-xxxx α3:33-xxxx 对所有试样都应用上面三张卡片,那么你会得到相对准确的值,具有比较高的可比性。 有时候你的试样里可能除了α1,α2,α3外,其中Ak#试样里还含有一个β相,这时候对于β相的卡片的选择非常重要,选择不好可能会对结果造成相当大的影响。有时候也可根据需要,去掉该相重新做。 有些卡片没有RIR值,这个时候就不能用这个方法了。如果实在需要,就自己查阅一下相关的资料找到一个RIR,或者自己求RIR。做法也很简单: 用纯的、结晶好的本相和α-Al2O3混匀研磨、扫描,然后计算各自最强峰的强度,得出RIR。 以前有人认为只比较衍射峰的强度(计数)就可以得出半定量的结果,其实这是不对的。我们得到的所谓的强度一般指的是衍射峰的绝对强度,比如3000或5000。这个数字除了能在统计学上对统计数据质量有一点保证外,再没有任何意义(这个数字越大说明数据越可靠)。 如果你认为同一张图里α1相和α2相的峰强度比较α1的大,就认为α1相的含量大,这是不对的。因为决定强度大小的因素除了量的多少以外还有很多,如物相的质量吸收因素,结晶程度,多重性因子等。如果把等量的quartz和αAl2O3均匀混合,测量发现quartz的峰要强出很多倍。 如果你认为α1相在图A1里的强度和A2里的比较A1里的大,就认为A1试样中α1相的含量大,这也是不对的。影响这个强度的因素也很多,如试样量的多少,扫描速度,射线量,干燥程度等都会影响到强度。我们做半定量分析一般是不会对试验提出过细的要求的,大多都是在一张满足做物相定性分析的图谱上去进行。而做定性分析对图谱的质量要求是较低的,1g试样或2g试样都可以,时间常数5s或20s也不会有什么区别,但这些因素都会对强度有很大的影响。 综合以上两点,说明RIR法半定量分析的科学严谨性,不是说半定量就可以很糊弄,它同样是科学的,只要我们用科学的态度和科学的方法。定量分析(quantitative analysis) 实际上是半定量分析方法的一个延伸。 首先用你最相信的方法测量一个或多个精确的定量分析结果,也可以事先精确配比一系列的试样: A1:α10%,β20%,y70% A2:…… A3:…… …… 把所有试样在普通的程序下扫描,然后按照半定量分析的方法选择合适的卡片,使其结果和事先配比的一致,记下该卡片号。以后你在对该系列的试样进行定量分析的时候,就可以把半定量结果当作定量结果使用。有时候可能找不到一组合适的卡片,就需要对已有卡片的RIR值进行修改,使之满足我们的要求。具体的命令行如次: custom → program settings → reference → allow database modification 打勾→ ok 选择卡片→ modify RIR → 修改 → ok 最后把打的勾去掉。制做pdf卡片的方法: custom → program settings → reference → allow database modification 打勾→ ok file → open *.caf reference pattern → add… → 输入卡片的相关信息(很多要素)→ ok →最后把打的勾去掉。注意:修改和制做pdf卡片是慎之又慎的事情,不能随意修改。最好能做一个专门的记录,内容包括被修(制做)改的卡片号码,修改前的内容,修改(制做)的日期,修改后的内容,以备恢复。 当然highscore也可以做更精确的Rietveld定量分析,很多测量结果可以达到0.001的精度,而且不用称量,不用配比,不用参考RIR,是真正的精确的无标定量分析法。有兴趣的朋友可以一起探讨。 最后恳请朋友们多提宝贵意见,指正错误和不足。
请问:双丙酮-D-葡萄糖的沸点为362℃,产品用色谱进行定量分析,可以用气相吗,如果可以的??可以,请给操作条件; 如果用液,请问用什么检测器??熔点110℃左右







 我要推广仪器
我要推广仪器
 下载APP
下载APP