液相色谱柱损耗率高?五大维护关键问题必看!
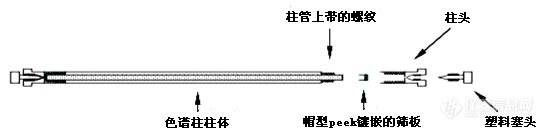
p style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "色谱柱技术始于上世纪50年代,随着填料和填充技术的发展,色谱柱技术日益成熟,功能也日趋完善,目前已被广泛应用于生命科学、环保、材料、食品、药物开发等领域。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "液相色谱柱在色谱分析系统中主要起着分离检测物质的作用,如同色谱系统的心脏,同时也是易损耗品。为了减少损耗,色谱柱的使用维护至关重要!/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "液相色谱柱使用过程常用问题包括色谱柱连接、色谱柱活化、色谱柱使用、色谱柱维护、色谱柱保存等。/span/pp style="text-align: center"img style="max-width:100% max-height:100% " src="https://img1.17img.cn/17img/images/202007/uepic/f8604747-9570-4f4c-ab4c-38392323be4a.jpg" title="1.png" alt="1.png"//pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun " /spanspan style="font-family: 宋体, SimSun " /span/pp style="line-height: 1.75em text-indent: 2em "strongspan style="font-family: 宋体, SimSun "1、色谱柱连接/span/strong/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "色谱柱安装方向/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "色谱柱安装应按照同一个方向连接使用,且需要按照色谱柱上的方向指示连接,strong尽量避免色谱柱反向连接!/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "常见色谱柱连接的问题主要有两种,安装色谱柱时管线伸出接头长度过长,使得螺纹拧入较浅,会导致密封性不好而漏液,进一步引起基线漂移或响应降低;反之,会在管线前段出现死体积,引起峰形展宽,灵敏度降低。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "理想的接头连接应具备以下特性:管线与接口之间无死体积;在超高压和高温下始终避免泄漏;优异的长期使用稳定性,防止管线滑动;简便易用。/span/pp style="text-align: center"img style="max-width:100% max-height:100% " src="https://img1.17img.cn/17img/images/202007/uepic/84c8701f-71fc-4530-a9f0-a1a71937ed61.jpg" title="2.png" alt="2.png"//ppbr//pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "管线的选择也非常重要,分析型液相系统最常用的规格是0.12和0.17mm内径的管线。更换管线时首先要确认当前管线的规格、并更换相同内径和长度的管线,否则会造成更换前后结果的不一致,因为管线体积会影响系统柱外体积,从而影响峰形和保留时间。/spanspan style="font-family: 宋体, SimSun text-indent: 2em " /span/pp style="line-height: 1.75em text-indent: 2em "strongspan style="font-family: 宋体, SimSun "2、反相柱活化平衡/span/strong/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "1) 首先,使用甲醇或乙腈冲洗约20 倍柱体积 。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "2)若流动相含有缓冲盐,使用与流动相中初始比例相等比例的超纯水和有机相冲洗过渡约20 倍柱体积,再用含缓冲盐的流动相平衡冲洗约20 倍柱体积或以上。 /span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "3) 若流动相不含缓冲盐,可直接用流动相平衡色谱柱,大约20 倍柱体积或以上。 /span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "4)当基线和压力平稳后测试,判断是否充分平衡以连续进样结果的重现为准。若不够可延长流动相的平衡时间。/span/pp style="line-height: 1.75em text-indent: 2em "strongspan style="font-family: 宋体, SimSun "3、反相柱冲洗保存/span/strong/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "1)使用50:50 甲醇或乙腈与水的混合溶液冲洗20-30 倍的柱体积;/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "2)使用纯甲醇或乙腈冲洗20-30倍柱体积;/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "3)储存之前将堵头紧紧密封在柱端接头上,以免填料变干。/span/pp style="line-height: 1.75em text-indent: 2em "strongspan style="font-family: 宋体, SimSun "4、反相色谱柱清洗再生/span/strong/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "清洗或反冲清洗反相色谱柱时,用以下溶剂至少各30倍柱体积冲洗色谱柱:/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "断开色谱柱与检测器的连接,将管线留在色谱柱末端,将其放入接收液体的烧杯中,先用不含缓冲液盐的流动相冲洗(水/有机相),然后用 100% 有机相(甲醇和乙腈)冲洗,检查压力是否回归正常,如果没有,再进行下一步操作。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "如压力没有回归正常,丢弃色谱柱或考虑用更强的条件清洗:75% 乙腈/25% 异丙醇、100% 异丙醇、100% 二氯甲烷 、100% 己烷。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun text-indent: 2em color: rgb(84, 141, 212) "值得注意的是,无论是使用己烷还是二氯甲烷,使用之前或恢复使用反相流动相之前必须用异丙醇进行冲洗。/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "关于色谱柱反冲/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "虽然色谱柱不应轻易反冲,但当明确知道超压来自颗粒物堵塞筛板或柱头污染时,反冲是最有效补救方法。反冲色谱柱可使颗粒物快速被冲出,此外还可快速冲出柱头强吸附污染物,柱子反冲后最好仍然正向连接使用。不过,反冲也会带来负面影响,如可能导致柱床松动、发生保留时间改变、小粒径的色谱柱反冲可能导致填料流出等。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "其中,可以反冲的色谱柱有:粒径大于2um的色谱柱(2.7、3、3.5、4、5μm等);而不可反冲的色谱柱有:粒径小于2um的色谱柱(1.8μm RRHD/RRHT;1.9μm Poroshell)。/span/pp style="line-height: 1.75em text-indent: 2em "strongspan style="font-family: 宋体, SimSun "5、色谱柱使用过程中常见问题/span/strong/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "液相色谱柱使用过程中最常见的问题包括pH值、温度、溶剂耐受、压力、样品等。色谱柱使用条件不得超出厂家建议的范围,包括最高压力,pH范围,水相耐受,柱温等。当测试条件接近色谱柱使用范围的极限值时,柱寿命会受影响。/span/pp style="text-align: center"img style="max-width:100% max-height:100% " src="https://img1.17img.cn/17img/images/202007/uepic/59a6513a-a6d8-46e8-9bbf-8de6be7224c5.jpg" title="3.png" alt="3.png"//pp style="line-height: 1.75em text-indent: 2em "br//pp style="line-height: 1.75em text-indent: 2em "span style="font-size: 20px "strongspan style="font-family: 宋体, SimSun "问题集锦/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "1、C18柱子如何调PH和温度以提高分离度呢?/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:通过调整pH和柱温优化分离度,这是方法开发中非常重要的手段。简单来讲,中性或不可电离化合物对pH变化不敏感。对于可电离化合物而言,可以通过调整流动相pH值,控制化合物电离状态来改变化合物的反相保留。降低pH可增大酸性化合物保留,而提高pH则可增加碱性化合物保留。通过调整pH改变化合物保留进而优化各个组分之间的分离度。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "通常提高柱温使得传质加快,保留也会降低,但是不同化合物保留对温度变化敏感程度不同,因此也可以通过调整柱温改变各个组分的保留时间来优化分离度。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "2、色谱柱总超压可能是什么原因呢?/span/strong/spanspan style="font-family: 宋体, SimSun " /span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:超压一般是液相流路内部包括色谱柱在内可能有堵塞。需要先做分段排查确定堵塞的部位,再根据堵塞部位排查引起堵塞的可能原因。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "如果是色谱柱堵塞,比较常见的原因有很多,如样品脏、基质复杂并且没有经过良好的预处理,或者预处理之后进入液相系统后又析出从而造成堵塞或污染(解决方法:加强样品预处理);色谱柱超压或超出pH范围使用导致填料碎裂,碎屑颗粒堵塞色谱柱(解决方法:根据测试条件选择合适色谱柱,避免超范围使用);仪器使用过程中部件磨损碎屑造成的堵塞(解决方法:及时更换受损部件)等等,都会引起系统色谱柱压力升高。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "3、C18柱子出峰时间拖后是什么因素影响?用一段时间出峰时间就拖后了,请问与流动相有没有关系?/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:液相色谱中影响化合物保留的主要因素包括:样品,色谱柱,流动相(流速,组成,比例等),柱温等。使用过程中发现保留时间漂移的话,需要从以下几个影响因素进行排查:可以先通过对比保留时间漂移前后相同条件下的压力曲线是否重现,从而初步排查可能的原因。若压力曲线不重现,首先确认测试条件是否有改动,检查流动相流速,组成,比例等是否改变,是否存在漏液或进气泡引起的流速和比例变化;对流动相组成变化敏感的样品和方法,应确保每次配制流动相的重现性;检查色谱柱是否堵塞污染;仪器控温是否准确等等,可能的原因比较多,具体原因需要进一步排查。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "4、色谱柱用什么流动相保存最好?用纯有机试剂是否容易干?/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:反相柱可以用HPLC级的甲醇或者乙腈保存,注意紧密连接堵头。正常情况下只要堵好堵头,溶剂是不容易干的。当然在保存溶剂中添加5%-10%的水,也没有问题。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "5、乙腈流动相总是容易聚合,有没有什么解决办法?/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:乙腈的聚合需要一定条件和时间,务必使用品质可靠的HPLC级溶剂,并且保证所使用溶剂尽可能新鲜。如果是放置保存比较久的乙腈溶剂,使用之前先过滤一下再用会有一定改善。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "6、小分子极性物质一般选用什么液相色谱柱?/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:可以先尝试用能够耐受高比例水相的柱子,提高流动相水相比例来增强保留。如果是可电离化合物,如酸性或者碱性化合物,可以在反相模式下先尝试通过调整流动相pH增大保留,酸性化合物需降低流动相pH,碱性化合物则提高流动相pH,根据pH条件选择可以耐受的色谱柱。如果调整pH后反相模式保留仍然很弱,您还可以考虑使用其他保留模式的色谱柱,例如HILIC柱,HILIC-Z,HILIC-OH5,或者纯硅胶的HILIC柱等等,也可以使用离子交换色谱柱或者正相色谱等。/span/pp style="line-height: 1.75em text-indent: 2em "span style="color: rgb(192, 0, 0) "strongspan style="font-family: 宋体, SimSun "7、柱子分离效果差了该怎么处理?/span/strong/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "答:导致色谱柱分离度降低的原因,主要是色谱柱柱效下降及色谱柱选择性发生改变。引起柱效下降的原因比较多,如果是连接不当造成的柱效损失,重新正确连接即可。如果是色谱柱使用中由于柱子污染引起的柱效下降或选择性改变导致的分离度降低,可以尝试对柱子进行清洗再生。如果是色谱柱本身的损伤引起的柱效下降分离度变化,这种通常是不可逆的,只能更换色谱柱,并且在后续使用新色谱柱的时候尽量避免各种损伤柱子的操作。/span/pp style="line-height: 1.75em text-indent: 2em "span style="font-family: 宋体, SimSun "br//span/pp style="line-height: 1.75em text-indent: 2em text-align: right "span style="font-family: 宋体, SimSun " /spanspan style="font-family: 宋体, SimSun text-indent: 2em " i本文根据安捷伦报告整理而成,欲了解更多内容,请点击链接观看视频: /i a href="https://www.instrument.com.cn/webinar/video_113123.html" target="_self"https://www.instrument.com.cn/webinar/video_113123.html /a/span/ppbr//p




























 我要推广仪器
我要推广仪器
 下载APP
下载APP