最近测量铅砷汞镉出现了一个问题http://simg.instrument.com.cn/bbs/images/default/em09501.gif做标准曲线的时候汞镉的吸光度减半。砷更离谱,预热半个小时灯能量始终不稳定,负高压接近600,标准曲线吸收值很低,线性不好,请各位老师帮我看看问题出现在哪里? 首先介绍一下基本情况http://simg.instrument.com.cn/bbs/images/default/em09511.gif仪器是耶拿ZEEnit 700 铅镉是用石墨炉做的,砷汞是用氢化物做的。在测量过程中,铅标准曲线的吸光度跟以前相比没有改变,线性良好(能达到0.999);做镉标曲时线性勉强说得过去,但吸光度减小;汞标曲线性不错,但吸光度值仅为原来一半;砷标准曲线每点吸光度值都很小而且不成线性,噪音较大。 针对以上问题我尝试了解决办法,但是效果不好。首先我清洁了石墨炉跟石墨管,并将石墨炉跟氢化物两边的6个石英窗拆下来清洗,从新做镉标曲,结果吸光度跟以前相比还是较小,然后我换了新的石墨管,并将原子化温度降低50℃,从新做镉标曲,结果吸光度并没有明显改善。清洗完石英窗,做汞标曲,汞灯能量稳定,负高压降低,但标准曲线各点的吸光度没有提高,我怀疑是标准品时间长了,于是从新配制标准品上机测试,结果还是原来吸光度的一半。最后再来做砷,由于清洗完石英窗,灯能量稳定了一点,负高压降到540左右,从做标准曲线,标曲各点的吸光度增大了一点,但不到原来吸光度的一半,线性勉强到0.995。 今天领导让我做仪器检出限,但是吸光度减半了,斜率必然减小,氢化物噪音比石墨炉大,砷汞肯定没法达到要求http://simg.instrument.com.cn/bbs/images/default/em09509.gif请各位老师帮我看看问题出在哪儿,我该怎么办?另外我想多学一些原子吸收相关的知识,能不能给我推荐几本书http://simg.instrument.com.cn/bbs/images/default/em09511.gif
西地碘用碘滴定液滴定的原理及化学反应方程式和注意事项
都晓得汞的残留效应很严重,而且因为样品中汞的含量很小,所以用ICP-MS分析汞其残留效应对分析结果影响很大,有时候甚至连标准曲线都拉不好,故实际工作中,很多人会用金溶液作为汞的稳定剂,对系统中汞的残留似乎有一定的清洗能力!
我们公司最近买了一台苏州青安公司的QM208B的冷[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]测汞仪。我根据HJ543的标准,使用高锰酸钾吸收液采集汞气体样品。然后使用10mL的反应瓶取5mL样品上机测试。发现数值很不稳定。测空白或者样品有时候峰值为0,有时候连续很多针峰值都固定在40左右,有时候同一个空白一针是峰值是0,下一针峰值又是40,再下一针又是0。数值一直突然升高,突然降低,完全没有规律。咨询工程师说我移取到反应瓶的液体量太多,我减少到2ml,问题依然没有解决。也试过用空气或用氮气做载气,依然是这个问题。请问这个问题可能是什么原因?要怎么解决?谢谢
贡献点FDA认证通过的天平室天平校正方法:用2套砝码,一套使用,一套备用,且都经过校验,每天对天平进行内校,采用5点法或7点法,称量砝码涵盖日常检验重量,校正要有记录,使用也要有记录。改天我把校正记录传上来。有问题我们再讨论。
按照中国药典,用原吸来做重金属,汞元素要单独消解吗?求大神指点,谢谢!铅镉砷汞的消解方法为:A法 取供试品粗粉0.5g,精密称定,置聚四氯乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内,进行消解(按仪器规定的消解程序操作)。消解完余后,取消解内罐置电热板上缓缓加热至红棕色蒸气挥尽,并继续缓缓浓缩至2~3ml,放冷,用水转入25ml量瓶中,并稀释至刻度,摇匀,即得。同法同时制备试剂空白溶液。汞的消解方法为:A法 取供试品粗粉0.5g,精密称定,置聚四氟乙烯消解罐内,加硝酸3~5ml,混匀,浸泡过夜,盖好内盖,旋紧外套,置适宜的微波消解炉内进行消解(按仪器规定的消解程序操作)。消解完全后,取消解内罐置电热板上,于120℃缓缓加热至红棕色蒸气挥尽,并继续浓缩至2~3ml,放冷,加20%硫酸溶液2ml、5%高锰酸钾溶液0.5ml,摇匀,滴加5%盐酸羟胺溶液至紫红色恰消失,转入10ml量瓶中,用水洗涤容器,洗液合并于量瓶中,并稀释至刻度,摇匀,必要时离心,取上清液,即得。同法同时制备试剂空白溶液。
[align=center][/align][align=center]对汞、砷、硒前处理方法改进的一点想法[/align][align=center]绿水青山VS金山银山战队[/align] 在环境监测中,对水样中汞、砷、硒等前处理有几种方法,包括《原子荧光法(HJ694-2014)》《水和废水监测分析方法(第四版)》《生活饮用水标准检验方法(GB 5749-2006)》等。对水样进行消解的目的有两个,一是破坏水中的有机物和悬浮物,减少干扰,二是使化合态元素转变成离子态,易于测量。在实验过程中,我对水样的消解处理有一些想法,在此提出来,与各位共同探讨。一.《原子荧光法(HJ694-2014)》中对汞的消解方法是量取5.0 mL混匀的样品于10 mL比色管中,加入1 mL盐酸-硝酸(1+1)溶液(稀王水)消解,然后定容。这就相当于对水样稀释了2倍,最后上机测量的结果应该乘以2。那么问题来了,一般来说,不稀释的水样与稀释2倍的水样测量结果并不严格呈2倍关系,并且,稀释倍数越大,造成的误差越大。 标准曲线浓度最高点一般设置为1ug/L,对于地表水、地下水来说,浓度一般不会超过1ug/L,测的值落在曲线范围里面。如果是这样,那么就没必要对水样进行稀释了。或者,对水样尽可能减少稀释倍数。与同行交流过程中,我有两个看法,一是取8mL 水样,然后取2 mL 稀王水定容到10mL,最后测的结果乘以系数1.25。二是取10mL水样,然后加入2mL稀王水,总共是12mL,最后测得结果乘以系数1.2。这两种方法的稀释倍数都比标准的稀释倍数要小,造成的误差应该也更小。 对于清洁的水样,比如地下水,也有版友说可以不经消解直接上机测量,或者加酸后上机测量。网址:1. [url]http://bbs.instrument.com.cn/topic/4009717[/url]2. [url]http://bbs.instrument.com.cn/topic/6026129[/url]3. [url]http://bbs.instrument.com.cn/topic/6834479[/url] 此外, 《生活饮用水标准检验方法(GB5749-2006)》中对水的消解方法是10mL水样加入1mL盐酸、0.5mL溴酸钾-溴化钾溶液,再加入1-2滴盐酸羟胺溶液,这样水样中大约为12mL,但是否乘以系数1.2,书上没有计算公式对此进行详细的阐述。二. 《原子荧光法(HJ694-2014)》中对砷、硒的消解方法是取50mL混匀样品,加入5mL硝酸-高氯酸混合液,再加入5mL盐酸,消解后定容到50mL容量瓶。如果测的样品数量较多,要消耗不少试剂,花的时间也比较多。如果将样品和试剂用量减半,达到节省药品的目的,而浓度不会有多大变化。 以上是本人的一点看法,欢迎各位共同探讨。
http://img.dxycdn.com/cms/upload/userfiles/image/2013/09/17/496868601_small.png根据清华大学官方网站消息,2013年9月13日,瑞典皇家科学院(Royal Swedish Academy of Sciences)宣布授予清华大学施一公教授2014年度爱明诺夫奖(Gregori Aminoff Prize),奖励他运用X-射线晶体学手段在细胞凋亡研究领域做出的突出贡献,奖金10万瑞典克朗(折合人民币93358.4元),颁奖典礼将于2014年3月31日在瑞典皇家科学院年会上举行。细胞凋亡(程序性细胞死亡)是在所有多细胞生物中起关键作用的基本生命过程,细胞凋亡的异常会导致严重病变,比如癌症、老年痴呆症等等,因此揭示细胞凋亡的分子机理可以加深科学家对这一基本生命过程的了解,并为开发新型抗癌、预防老年痴呆的药物起提供线索。爱明诺夫奖官方新闻稿提到,作为细胞凋亡机制研究的一部分,施一公的蛋白质晶体学研究不仅能让研究者深入了解蛋白质的三维结构,还能让他们详细了解蛋白质调节系统的详细机制。除此以外,施一公团队在生物学其他领域也提出了诸多开创性见解,例如,他的研究小组曾经确定了一组与早衰相关的跨膜酶,该酶在阿尔茨海默症的发生发展中起到了一定作用。施一公此次获得爱明诺夫奖,是该奖设立35年来,首次颁给中国科学家。对于施一公来说,是实至名归。施一公是中国著名的结构生物学家,长江讲座教授,国家杰出青年基金获得者,“千人计划”首批国家特聘专家,现任清华大学教授,生命科学院院长,普林斯顿大学教授;其领导的实验室主要运用X-射线晶体学,结合其它生物物理和生物化学方法研究生命科学的基本问题,在细胞凋亡调节机制、生物大分子机器组装与功能、重要膜蛋白结构与机理三个主要研究领域做出了重要的原创贡献。施一公2013年当选为美国艺术与科学院外籍院士,美国科学院外籍院士,成为美国双院外籍院士。瑞典皇家科学院爱明诺夫奖瑞典皇家科学院创建于1739年,以其专设的诺贝尔奖评选委员会而闻名世界。自1901年起,瑞典皇家科学院就开始负责每年的诺贝尔物理学奖和化学奖的评选,自1968年起,又加入了纪念阿尔弗雷德·诺贝尔瑞典银行经济学奖(诺贝尔经济学奖)的评选。除诺贝尔奖外,瑞典皇家科学院还负责评选克拉福德奖、肖克奖等国际性大奖。爱明诺夫奖同诺贝尔化学奖一样,是属于瑞典皇家学院颁发的国际类奖项,设立于1979年,用以奖励世界范围内在晶体学领域做出重大贡献的科学家,每年颁发给不超过3名科学家,个别年度空缺。本年度的爱明诺夫奖只有施一公一人获奖。
食品中重金属及有害元素的测定知识点——汞 (一)基本原理及方法要点 氢化物原子荧光光度法:检出限0.03μg/L,标准曲线最佳线性范围0~60μg/L。 冷原子吸收光谱法:最低检出浓度,压力消解法为0.4μg/kg,其他消解法为10μg/kg。比色法为25μg/kg。 (二)总汞 1.氢化物原子荧光光谱法 (1)原理(见GB/T 5009.17-2003)。 (2)样品消解:可选择高压消解法或微波消解法(见GB/T 5009.17-2003)。 (3)测定:配制低浓度标准系列(适用于一般样品测定)和高浓度标准系列(适用于鱼及含汞量偏高的样品测定)。根据情况任选浓度测定方式测量或仪器自动计算结果方式测量。 2.冷原子吸收光谱法 (1)原理(见GB/T 5009.17—2003)。 (2)样品预处理(见GB/T 5009.17—2003)。 (3)测定:分别吸取样液和试剂空白液各5.0ml置于测汞仪的汞蒸气发生器的还原瓶中,分别加入1.Oral还原剂氯化亚锡(100g/L),迅速盖紧瓶塞,随后有气泡产生,从仪器读数显示的最高点测得其吸收值,然后,打开吸收瓶上的三通阀将产生的汞蒸气吸收于高锰酸钾溶液(50g/L)中,待测汞仪上的读数达到零点时进行下一次测定。并求得吸光值与汞质量关系的一元线性回归方程。将所测得其吸收值,代入标准系列的一元线性回归方程中求得样液中汞含量。 3.双硫腙比色法 (1)原理:样品经消化后,汞离子在酸性溶液中可与二硫腙生成橙红络合物.溶于三氯甲烷.与标准系列比较定量。 (2)样品消化(见GB/T 5009.17—2003)。 (3)测定:取不同样品的消化液(全量).加20ml水。在电炉上煮沸10分钟.除去二氧化氮等,放冷。于样品消化液及试剂空白液中各加高锰酸钾溶液(50g/L)至溶液呈紫色,然后再加盐酸羟胺溶液(200g/L)使紫色褪去,加2滴麝香草酚蓝指示液.用氨水调节pH,使橙红色变为橙黄色(pHl~2)。定量转移至125ml分液漏斗中。吸取0、0.5、1.0、2.0、3.0、4.0、5.0、6.0μl汞标准使用液(相当于0、0.5、1.0、2.0、3.0、4.0、5.0、6.0μg汞)。分别置于125m1分液漏斗中.加10ml硫酸(1:19),再加水至40ml,混匀。再各加1ml盐酸羟胺溶液(200g/L),放置20分钟,并时时振摇。于样品消化液、试剂空白液及标准液振摇放冷后的分液漏斗巾加5.Oml二硫腙使用液.剧烈振摇2分钟,静置分层后.经脱脂棉将三氯甲烷层滤入lcm比色杯中,以三氯甲烷调节零点,在波长490nm处测光度.标准管吸光度减去零管吸光度,绘制标准曲线。 (三)甲基汞的测定 1.气相色谱法(酸提取巯基棉法) (1)原理:试样中的甲基汞。用氯化钠研磨后加入含有Cu2+的盐酸(1:11),(Cu2+与组织中结合的CH3Hg交换)完全萃取后。经离心或过滤。将上清液调试至一定的酸度。用巯基棉吸附,再用盐酸(1:5)洗脱,最后以苯萃取甲基汞.用带电子捕获鉴定器的气相色谱仪分析。 (2)巯基棉制备:(见GB/T50(5009.17—2003) (3)色谱柱:内径3mm.长1.5m的玻璃柱.内装涂有7%(m/m)丁二酸乙二醇聚酯(pEGS)或涂1.5%(m/m)OV-17和1.95%F-1或5%(m/m)丁二乙酸二乙二醇酯(DFGS)固定液的60~80目chromosorb W AW DMCS。 2.冷原子吸收法(酸提取巯基棉法) (1)原理:试样中的甲基汞,用氯化钠研磨后加入含有Cu2+的盐酸(1:11),(Cu2+与组织中结合的CH3Hg交换)完全萃取后,经离心或过滤,将上清液调试至一定的酸度.用巯基棉吸附,再用盐酸(1:5)洗脱,在碱性介质中用测汞仪测定,与标准系列比较定量。 (2)分析步骤:称取1.00~2.00g去皮去刺绞碎混匀的鱼肉(称取5g虾仁,研碎),加入等量氯化钠,在乳钵中研成糊状,加入0.5ml氯化铜溶液(42.5g/L),轻轻研匀。用30ml盐酸(1:11)分次完全转入100ml带塞锥形瓶中.剧烈振摇5分钟,放置30分钟(也可用振荡器振摇30分钟),样液全部转入50ml离心管中,用5ml盐酸(1:11)淋洗锥形瓶,洗液与样液合并,离心10分钟(转速为2000r。/rain)。将上清液全部转入100n11分液漏斗中,于残渣中再加10ml盐酸(1:11),用玻璃棒搅拌均匀后再离心.合并二份离心溶液。加入与盐酸(1:11)等量的氢氧化钠溶液(40g/L)中和,加1~2滴甲基橙指示液,再调至溶液变黄色,然后滴加盐酸(1:11)至溶液从黄色变橙色,此溶液的pH在3.0~3.5范围内(可用pH计校正)。将塞有巯基棉的玻璃滴管接在分液漏斗下面,控制流速约为4~5ml/min;然后用pH3.O~3.5的淋洗液冲洗漏斗和玻璃管,取下玻璃管.用玻璃棒压紧巯基棉,用洗耳球将水尽量吹尽,然后加入1ml盐酸(1:5)分别洗脱一次.用洗耳球将洗脱液吹尽,收集于10ml具塞比色管中。洗脱液收集在10ml具塞比色管内,补加铜离子稀溶液至10一ml。再吸取2.0ml此溶液.加铜离子稀溶液至10ml。 另取12支10ral具塞比色管,分别加入5ml铜离子稀溶液,然后加入0、0.20、0.40、0.60、0.80、1.0ml甲基汞标准使用液各两管,各补加铜离子稀溶液至10ml(相当于0、0.020、0.040、0.060、0.080、0.10μg甲基汞)。将试样及汞标准溶液分别依次倒入汞蒸气发生器中,加2ml氢氧化钠溶液(400g/L)、15ml氯化亚锡溶液(300g/L),通气后,记录峰高或记录最大读数,与绘制标准曲线比较。
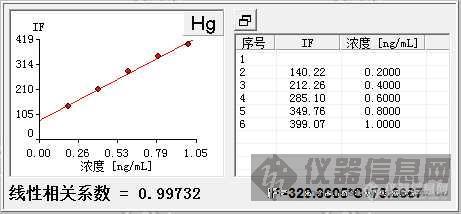
http://ng1.17img.cn/bbsfiles/images/2014/12/201412041652_525914_2903458_3.jpghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412041653_525915_2903458_3.jpg普析原子荧光做汞时曲线拟合零点能达到99.7%,而过零点曲线只有98.4%,标样是现配的,线曲是仪器自动稀释做的,标准空白和标样溶剂都是1%硝酸,为什么会出现这种情况呀,求解。。。
各位老师,近日在研读《药典》2015年版四部时发现,2321铅、镉、砷、汞、铜测定的第一法中,砷的测定规定使用氢化物法,汞的测定规定使用冷蒸气吸收法,如果我使用原子荧光进行砷、汞的测定算不算是方法偏离?如果严格按规定,到底能不能用原子荧光法进行砷、汞的测定?请大家不吝赐教!谢谢
请教为什么原子荧光分析汞不稳定?
样品前处理时,有沉淀析出,要用滤纸过滤后再进样,是先定容再过滤,还是先过滤再定容?请详细说明你的理由。

[size=18px][font=宋体][b]传染病的流行有三个环节:传染源、传播途径、易感人群。一是传染源是首要环节[/b],没有传染源就没有流行,从源头切断传播,事半功倍:需要封城、封路、禁止聚会、停止公共交通等措施,避免传染源的四处播散,减少接触传染源的机会;[b]二是传播途径[/b],目前来看呼吸道是主要的传播途径,直接接触也有可能,如接触了病人后揉眼,所以戴口罩,勤洗手等来减少患病机会,其他血液、粪便等同样具有传播途径;[b]三是易感人群[/b],容易感染新型病毒的人,目前新型病毒肺炎患者多为40-60岁,因此中年人尤其注意,有肝病、糖尿病、口服免疫抑制剂等免疫力低下的人群也应注意。老年人抵抗力低弱,更容易感染,更加需要注意并预防。[/font][b]面对新型冠状病毒肺炎我们应淡定面对,不恐慌。[/b][font=宋体]目前也是流感病毒及其他呼吸道病毒感染的高发季节,这些呼吸道病毒感染均可以出现发热,因此出现发热后莫要恐慌,积极就医,发热不代表就是新型病毒肺炎。[/font][font=宋体][b]团结一致,淡定应对,共抗病毒。主动从正规渠道学习关于新型冠状病毒感染的肺炎的知识,不信谣,不传谣,加强锻炼身体,提高身体抵抗力。[/b][/font][/size]
12V开关电源 X射线光电子能谱分析X射线光电子能谱法(X-ray Photoelectron Spectrom-----XPS)在表面分析领域中是一种崭新的方法。虽然用X射线照射固体材料并测量由此引起的电子动能的分布早在本世纪初就有报道,但当时可达到的分辩率还不足以观测到光电子能谱上的实际光峰。直到1958年,以Siegbahn为首的一个瑞典研究小组首次观测到光峰现象,并发现此方法可以用来研究元素的种类及其化学状态,故而取名“化学分析光电子能谱(Eletron Spectroscopy for Chemical Analysis-ESCA)。目前XPS和ESCA已公认为是同义词而不再加以区别。XPS的主要特点是它能在不太高的真空度下进行表面分析研究,这是其它方法都做不到的。当用电子束激发时,如用AES法,必须使用超高真空,以防止样品上形成碳的沉积物而掩盖被测表面。X射线比较柔和的特性使我们有可能在中等真空程度下对表面观察若干小时而不会影响测试结果。此外,化学位移效应也是XPS法不同于其它方法的另一特点,即采用直观的化学认识即可解释XPS中的化学位移,相比之下,在AES中解释起来就困难的多。1 基本原理用X射线照射固体时,由于光电效应,原子的某一能级的电子被击出物体之外,此电子称为光电子。如果X射线光子的能量为hν,电子在该能级上的结合能为Eb,射出固体后的动能为Ec,则它们之间的关系为: hν=Eb+Ec+Ws 式中Ws为功函数,它表示固体中的束缚电子除克服各别原子核对它的吸引外,还必须克服整个晶体对它的吸引才能逸出样品表面,即电子逸出表面所做的功。上式可另表示为: Eb=hν-Ec-Ws 可见,当入射X射线能量一定后,若测出功函数和电子的动能,即可求出电子的结合能。由于只有表面处的光电子才能从固体中逸出,因而测得的电子结合能必然反应了表面化学成份的情况。这正是光电子能谱仪的基本测试原理。
[font=宋体]发帖人:东北虎[/font][font=宋体]链接:[/font]https://bbs.instrument.com.cn/topic/4196[b][font=黑体]问题描述:[/font][/b][font=宋体]用原子荧光测定土壤中的汞但是仪器漂移严重,原子荧光为北京海光[/font]AFS2202[font=宋体],采用的灯是高性能空心阴极灯,方法是《岩石和矿石分析规程》([/font]DZG 93-01[font=宋体]),这是什么原因?[/font][b][font=黑体]解答:[/font][/b][font=宋体]原子吸收([/font]AFS[font=宋体])分析汞存在热漂移和灯漂移问题,确实经常出现,因此建议对土壤中汞的测定,推荐《土壤和沉积物[/font] [font=宋体]总汞的测定[/font] [font=宋体]催化热解-冷原子吸收分光光度法》([/font]HJ 923-2017[font=宋体])。对[/font]AFS[font=宋体]测定汞存在的漂移,可以采用以下措施:一是大电流如[/font]30 mA [font=宋体]预热小电流如[/font]15 mA[font=宋体]分析,预热时间要足够长如[/font]1 h[font=宋体]。二是实验室温度和还原剂溶液保持稳定,最好保持实验室门窗关闭,温度相差[/font]5[font=宋体]℃,仪器的灵敏度可能相差数倍;同时中途不要更换硼氢化钾还原剂溶液,否则重新建立标准曲线再分析实验空白和样品上机液。三是汞空心阴极灯漂移问题,摸索得出,每测试一定数量样品如[/font]15[font=宋体]个,再次测定标准曲线溶液中间点或合适浓度样品,如果再测信号值和首次测定信号值的误差大于[/font]5%[font=宋体],再测定后续样品时调低间隔样品数量如[/font]10[font=宋体]个,并调整负高压(理论依据:[/font]AFS[font=宋体]的信噪比不随负高压的调整而变化)再测该样品的信号值,直至测定信号值和首次信号值相对偏差小于[/font]5%[font=宋体],然后再测后续样品。对低浓度汞样品,最好采用玻璃或石英材质比色管、上机小管;土壤样品混匀和盛装时,最好保存于纸质袋中,再用塑料密封袋套封,避免样品间交叉污染。[/font][font=宋体]此外,鉴于部分土壤样品(含标准物质)中汞的含量很低,必须确保汞实验空白足够低。[/font]
本书论述在测量仪器的计量校准(或检定)过程中普遍遇到的问题,由于标准自身存在不确定度,影响到对被检仪器检定结论的正确性,即有可能将原本不合格的仪器误判为合格,从而造成用户风险;或者将原本合格的仪器误判为不合格,从而造成厂家风险。本书对这两类进行了深入的分析和定量的计算。在此基础上,提出了多种解决的方案。 本书论述深入浅出,提供有一定深度的理论背景分析,同时提供大量图表,以方便在实际工作中查阅和使用。主要供从事测量仪器的制造、使用、校准和检定人员,以及计量管理部门的技术干部和大专院校相关专业的师生阅读和参考。第1章 预备知识 1.1 理解仪器的指标 1.2 何谓测试不确定度比第2章 检定结论的总体风险 2.1 总体用户风险的产生原因 2.2 总体厂家风险的产生原因 2.3 常见检定条件下的总体风险概率 2.4 对两种总体风险的简单评论 2.5 小结第3章 限制总体用户风险的对策 3.1 用保护带压缩总体用户风险 3.2 总体用户风险攀比高tur准则 3.3 保护带对总体厂家风险的影响 3.4 确定压缩因子k的其他几种方法 3.5 根据不同方法确定的k因子 3.6 小结第4章 单 台仪器检定结论的风险 4.1 单台仪器检定结论用户风险的产生原因 4.2 单台仪器检定结论的厂家风险 4.3 典型检定条件下单台仪器检定的风险概述. 4.4 小结第5章 限制单个检定结论用户风险的对策 5.1 用限制总体风险的方法来限制单台仪器检定结论风险 5.2 作者建议采用的限制单台仪器检定结论用户风险的方法 2.3 小结第6章 具有被检仪器先验知识时单台仪器检定结论的风险 6.1 具有被检仪器先验知识时单台仪器检定结论的用户风险 6.2 具有被检仪器先验知识时对单台仪器检定结论的厂家风险 6.3 典型条件下的用户风险 6.4 具有被检仪器计量特性先验知识时典型条件下的用户风险值 6.5 已知被检仪器具有高斯分布特征性时对用户风险的限制方法 6.6 小结第7章 单台仪器检定结论的风险权衡 7.1 风险权衡问题的提出 7.2 两类风险之间的关系 7.3 用户优先因子和风险的定量权衡 7.4 用户优先因子和保护带的关系 7.5 小结第8章 下检定结论时的一些实际考虑 8.1 出具检定结论的可能方式 8.2 对“另一个测试限”的选择 8.3 典型条件下的允差带“扩展因子” 8.4 小结
gonglex说:刘老师您好,向您请教个问题。我在检定汇松MB-530酶标仪时,(1)此机型属于I类,在检定波长时应该怎么操作呢?按照规程用检定分光光度计的干涉滤光片,但是片子尺寸大啊,放不进去啊,如何办呢?(2)在405和450波长下,1.5这个点上超差,而在其它波长时都是合格的,不知道是什么原因了? 望刘老师赐教!
因为需要用硫代硫酸钠滴定样品中的碘含量,由于不知道样品中碘的具体量,也不能估计硫代硫酸钠的用量,所以在滴定反应一开始即加入淀粉用于判断终点,将样品滴至蓝色消失,放置一段时间后又出现蓝色,怀疑由于淀粉对碘的吸附乃至滴定不完全,又用硫代硫酸钠滴到终点,可相同情况又出现,之后只好再滴加硫代硫酸钠,直到样品不再出现蓝色,请问这样操作的过程后,将硫代硫酸钠每次所用量加和,是否能作为准确值进行计算(注:样品溶液为酸性环境)
谁知道有什么仪器可以测量0到-50℃共晶点的仪器设备?感谢推荐个品牌型号
原子荧光测生活中水硒 汞 砷,在计算含量的时候定容体积应该填写多少呢,是写10ml还是加上后来添加试剂的体积?
用硫代硫酸钠滴定碘伏消毒剂,当溶液变成浅黄时加入淀粉溶液,不变色,没有蓝色出现。以前滴定碘类消毒剂偶尔也会遇见这种情况。请教各位大侠这是怎么回事?我滴定的这个碘伏溶液是聚醇醚碘,由脂肪醇聚聚氧乙烯醚、碘、碘化钾、醋酸配一起络合而成。我在想会不会是配制的碘伏溶液的某些因素影响到淀粉变蓝?各位大侠有知道的吗?跪求答案。
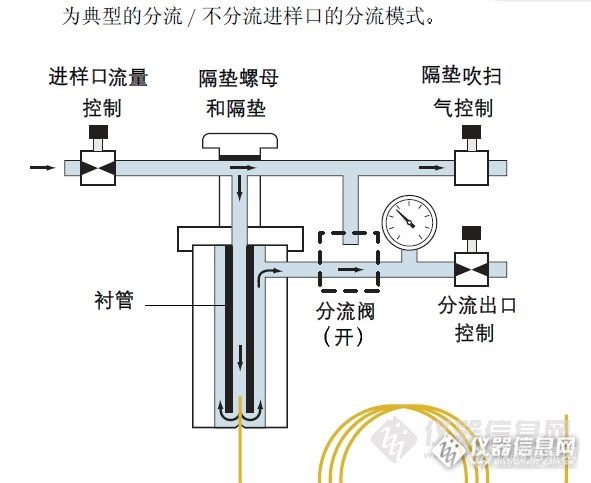
(本文仅提供刚刚入分析行业的同学借鉴, 有不到之处,请各位老师多提宝贵意见)一提到化工分析,大家就想到瓶瓶罐罐,每天就是滴滴摇摇,洗洗刷刷,但现在随着时代的变化,基本功要求越来越少,当年的机械分析天平被电子天平代替;打鸡蛋一样的甩瓶子被自动滴定仪代替。仪器分析比重越来占越大。所以来谈谈怎样来学习仪器分析。 首先我们要做到这个前提:你能熟练的操作仪器!切记! 其后要知道仪器分析理论,工作原理,了解和熟悉相关仪器的构造。 我以色谱为例: 色谱有气相色谱,液相色谱,离子色谱,毛细管色谱等等。但千变万化,他们的原理都是一样的,就像河里有一堆大小石子,用水流来冲,小的因为质量和与河床摩擦力的原因,移动的速度比较快,而大的石头移动的比较慢,我们在下游某个截面上数冲下的石头,经过一定的时间,可以分开每个规格的石头各有多少。以此类推:放一堆石子就是我们色谱进样,水流就是我们的流动相,区别只是气相色谱是气体,液相色谱,离子色谱是液体,毛细管色谱靠的是高压电流和压力相结合,目的都是给他一个流动的动能。在冲刷过程,河床的影响就等同我们的色谱柱,不管什么样的色谱柱,目的都是极力的拉住所有的物质,最终使不同物质分开,而在某个截面上数下的石头就是检测器干的事情,区别只是光还是电。(注:光指紫外检测器等,电指FID,TCD等根据电流变化设计的检测器)。原理知道了,我们在来具体分析每个部分组成,他们各自的作用是什么?以气相分流/不分离进样口为例:http://ng1.17img.cn/bbsfiles/images/2016/07/201607290014_602455_1637344_3.jpghttp://ng1.17img.cn/bbsfiles/images/2016/07/201607290014_602455_1637344_3.jpg我们拆开可以看到:隔垫螺母,隔垫,O型圈,衬管,分流平板,石墨垫,色谱柱,再加上加热模块。不同的色谱配备了不同的气路控制装置。知道这些,在以后工作中遇到故障就可以有针对性的解决。如进样口压力不足,首先检查漏气,看有关密封方面的,如隔垫,O型圈,石墨垫是不是损坏。再查色谱柱是不是出问题了。最后检查气路控制是不是失灵了!再如有鬼峰,查进样口污染,看衬管是不是脏了,分流平板是不是有问题,我见过2年没有换过的分流平板,原先金黄色的分流平板变的全部黑色。,同时考虑有什么可以进入进样口的,如载气。熟悉了各部件,对你解决工作中的问题帮助巨大! 我们在熟悉一个仪器时,还要考虑这个仪器的一些部分在另外的仪器上也有使用,因为仪器很多都是相通的,比如紫外检测器。最常见的是:紫外分光光度计。这个组件在液相色谱,毛细管色谱上都有使用。有关紫外检测器出现的问题,下次遇到别的仪器出类似问题的时候,我们有个参考,能很快的判断出来。 勤动手,这个不是让你不管三七二十一,出了问题,自己拿个工具就拆,如果这样的话,多出一个二个零件的事情概率很高。这个勤动手是让你,在维修工程师维修的时候,注意看,多用手机拍点资料,帮工程师打打下手,在工程师在场的情况下,先外围,后核心的尝试拆解和组装,感受螺丝,部件到位的手感。 勤动嘴,多问,不管是工程师还是普通分析工,达者为师。多访问专业论坛,比如仪器信息网等等。不懂的东西可以发在论坛上求助,讨论! 有老师补充的,欢迎回帖!
原子荧光怎样才能测准样品中的铅(整理)做原子荧光测定样品中的铅时,标液中荧光值出现今天高,明天低的现象,而且有时数值差异很大。请教:怎样配制载流和还原剂中的酸碱度才能使测定结果较好。?1.原子荧光测定,我做的比较多的是汞砷硒,样品多为土壤,植株,水系,畜产品。在检测过程中,发现原子荧光试剂和配置的溶剂都比较重要,大家都购买的时候,不仅要注意纯度,更要检测一下试剂的空白。首先说明,我使用得试剂都是国产,从上海国药集团试剂公司购买。盐酸:这个常用,必须是GR,建议检测10%盐酸汞空白。还有就是盐酸不能混瓶使用,就是一瓶用了一大半,把它和另一瓶合用。硼氢化钾(钠),它关系到砷硒的检测,纯度为95%(国产)。有时候,作汞没有问题,作砷硒就出不来信号,仔细看看仪器气液分离树那里没有气泡产生,没有反应,可能就是硼氢化钠失效了,无法产生大量的氢气。请注意,该试剂怕潮,平时最好放置在干燥皿内。我发现有一种小瓶100g的好用,黄白包装的,一直用到完都好用。有一种500g大瓶装小半瓶,也是100克的,不好用,刚开始没事,后来效果就差了。溶液当天配置,冷藏最多用3天,我一般现配现用。氢氧化钾(氢氧化钠)用来保护硼氢化钠,GR,我一般配置还原剂后加几块固体,现配先用不需要加很多如0.5%,加一点就够了。但是装还原剂最好使用小口瓶,和空气接触面小的寿命长一点,受污染也小。还原剂浓度由试样来定,载流酸浓度由还原剂浓度来定,最后废液呈酸性。例如单独测定汞,还原剂可以配置成0.2%硼氢化钠,酸也就0.5%够了;如测定砷,硼氢化钠需要配置到2%,酸就要到5%。一句话,只要硼氢化钠可以满足标准曲线最高点,把溶液浓度放宽一倍即可,而酸只要能够保证废液呈酸性。废液为何要呈酸性?如果不是酸性,硼氢化钠会在管道压力最低处沉积,而后堵塞,当你看到管路内液体要停顿好长一段时间再冲上去的时候,下一次它们就冲到你脑门上了,呵呵。硫脲:AR就可以了,我买了500g一大瓶,慢慢用。Vc,国产的只有AR,不过纯度也够食用了。开瓶后不能久存,会被空气氧化的。一般我是把硫脲和Vc按照1:3混合,用碾钵碾碎,在测定前半小时用最小号药匙每个样品和标准曲线加一点点,充分振荡还原,效果明显而且快,呵呵就是费试剂和野蛮,不知有没有同道。一般作硒我也使用Vc还原,感觉快,比盐酸好。标准溶液:购买。汞中间液(和保护液)我配的是0.02%重铬酸钾(0.5%盐酸)溶液,试样消解完了就用它来定容,测定砷硒再加上面讲的固体还原剂,简单。最好一天内消解测定,注意自身防护。2.一般我是把硫脲和Vc按照1:3混合,用碾钵碾碎,在测定前半小时用最小号药匙每个样品和标准曲线加一点点,充分振荡还原,效果明显而且快,呵呵就是费试剂和野蛮,不知有没有同道。[/quote]感觉不严谨,应该是配成溶液,加入相同体积溶液用药匙不准
有0,.1mol硫代硫酸钠滴定17%一元过氧乙酸时,先加5ml硫酸(1+9),然后加3滴钼酸铵,然后加入10ml稀释后的过氧乙酸,然后加10ml碘化钾(100g/L),在阴暗处放置10min,由于过氧乙酸含量较高,所以每次碘量瓶中有大量碘析出,滴定结果每次消耗的硫代硫酸钠体积都基本不一样(同一容量瓶里取出的3个样,其结果也都不太一致),这是由于滴定速度问题还是摇动速度问题,不明所以,请大神指点
样品中含有伯胺(且仅有这一个氨基),客户给的方法为”使用高氯酸做为滴定液,非水电位滴定法测定含量,读取两个电位突跃点之间消耗滴定液的体积,以该体积计算含量。”个人理解:1、两个突跃点应分别是空白溶剂与供试品与滴定液反应达到终点时的电位突跃;2、在供试品溶液中,应该是样品先与滴定液发生反应,空白溶剂随后再与滴定液,那么两个突跃点之间的体积应该相差很小才对。那么,上面的假设就是错误的。现在正矛盾着呢?哪位老师能够帮助解决一下呢?谢谢了
固定源废气中汞的测定 标准方法是汞的测定 冷原子吸收法(HJ534-2009),计量认证能否能应用《空气和废气监测分析方法》中的原子荧光法,原子荧光法用的是滤筒采样,滤筒只能采集烟道废气颗粒中的汞,还是也能收集废气气体中的汞?

[align=center][font=黑体][size=18pt]烷基汞仪器分析新方法[/size][/font][/align][font=楷体_GB2312][size=12pt] ([/size][/font][font=楷体_GB2312][size=12pt]老兵)[/size][/font][b][font=黑体]摘[/font] [font=黑体]要[/font] [/b][font=楷体_GB2312]本文介绍了环境样品中水体、土壤和沉积物的甲基汞常用检测方法,讨论了各检测方法的优势和缺欠,并展望了甲基汞检测方法未来的发展和应用前景。目的是建立方便、快捷、灵敏度高的方法,应用于日常检测中烷基汞(甲基汞、乙基汞)的测定,提升国内在检测烷基汞(甲基汞、乙基汞)方面的技术水平。以满足有效检出烷基汞(甲基汞、乙基汞)的环境管理要求和显著提高工作效力。[/font][b][font=黑体]关键词:烷基汞[/font] [font=黑体]测试[/font] [font=黑体]方法[/font][font=黑体][size=14pt]1 [/size][/font][font=黑体][size=14pt]烷基汞及其来源[/size][/font][/b][font=宋体][size=12pt] 烷基汞(甲基汞、乙基汞)是一类剧毒并且有强致癌作用的有机金属化合物,其毒性远大于无机汞的毒性。烷基汞中甲基汞毒性最强,环境中任何形式的汞(金属汞、无机二价汞和烷基汞等)均可在一定条件下转化为有剧烈毒性的甲基汞,称为汞的甲基化。甲基汞又分为一甲基汞(氯化甲基汞、碘化甲基汞等)和二甲基汞。汞的甲基化反应主要发生在水体中,并在食物链中富集,浓度逐渐增大。水中胶体颗粒、悬浮物、泥土颗粒、浮游生物等能吸附汞,而后通过重力作用沉降进入底泥,底泥中的汞在微生物的作用下可转变为一甲基汞或二甲基汞。甲基汞[/size][/font][font=宋体][size=12pt](MeHg)[/size][/font][font=宋体][size=12pt]能溶于水,又可从底泥返回水中,[/size][/font][font=宋体][size=12pt]作为高脂溶性的甲基汞,极易被生物体吸收和利用,且在生物体内分解缓慢(半衰期约为70d),易发生生物富集,如鱼类对甲基汞的富集系数可达鱼类可高达1×10[sup]5[/sup]~1×10[sup]7[/sup]。通过食物链的传递放大作用,甲基汞严重威胁人类的健康与安全。其中最为典型的例子就是日本熊本县水俣湾地区的居民因长期食用受甲基汞污染的鱼贝类而引起的慢性甲基汞中毒,即水俣病。这次世界历史上首例重大重金属污染事件,引起了全球研究者对汞污染问题的广泛关注。[/size][/font][b][font=黑体][size=14pt]2 [/size][/font][font=黑体][size=14pt]传统的烷基汞测试方法[/size][/font][/b][font=宋体][size=12pt] 随着汞污染问题研究的不断深入,烷基汞检测方法的研究也不断发展起来。国内对烷基汞检测方法的研究起步较晚,早期常用的分析方法是以巯基棉富集一[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法为代表的方法,并于1993年和1997先后颁布了水质和环境中烷基汞测定的[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法,2009年中国环境监测总站又在下发的《集中式生活饮用水地表水源地特定项目分析方法》中增加了水质甲基汞测定的高效液相色谱-原子荧光法。由于环境中的甲基汞是通过汞的甲基化过程产生的,这一过程主要发生在沉积物及水体中,产生的一甲基汞极易溶于水体,只有少量的二甲基汞散逸到大气中,之后立刻被光解为甲烷、乙烷和汞,因此现有的检测手段,通常难以检测出大气中的甲基汞。[/size][/font][font=黑体][size=12pt]2.1[/size][/font][font=黑体][size=12pt][url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[/size][/font][font=宋体][size=12pt] 水中烷基汞的前处理方法有两种,一种是采用巯基纱布和巯基棉二次富集的前处理方法。图1为便携式(第一次富集用)的巯基纱布旋转富集装置,图2为巯基棉管(第二次富集用)吸附装置。水样先经巯基纱布富集后,用2mol/L的盐酸溶液洗脱后再用巯基棉进行二次吸附。而沉积物和污泥样品则须先用盐酸-硫酸铜溶液浸提,浸提液再按水样的巯基纱布和巯基棉二次富集方法进行前处理。将二次吸附后的巯基棉置于微型萃取管用苯萃取后,用[url=https://insevent.instrument.com.cn/t/Mp]气相色谱仪[/url](ECD检测器)测定苯相。[/size][/font][font=宋体][size=12pt][img=,630,339]http://ng1.17img.cn/bbsfiles/images/2014/07/201407160009_506821_1634717_3.jpg[/img][/size][/font][font=宋体][size=12pt][font=宋体] 第二种方法是采用巯基棉管的一次富集法。该巯基棉管为内径5~8 mm,长100 mm,一端拉细的玻璃管中填充0.1~0.2 g巯基棉纤维,使用前用20 mL无汞蒸馏水润湿膨胀,然后接在分液漏斗的放液管上使用。取1L水样置于2L分液漏斗中,加入硫酸铜溶液、盐酸溶液或氢氧化钠溶液,调pH为3-4,接琉基棉管,让水样流速保持在20~25m l./min,待吸附完毕,加人NaCl- HCl解析液,将琉基棉上吸附的烷基汞解析到离心管用甲苯(苯)萃取,取有机相进行色[/font][/size][/font][font=宋体][size=12pt]谱测定。[/size][/font][font=宋体][size=12pt]其标准色谱图如下:[/size][/font][font=宋体][size=12pt][img=,606,319]http://ng1.17img.cn/bbsfiles/images/2014/07/201407160008_506820_1634717_3.jpg[/img][/size][/font][font=宋体][size=12pt] 甲基汞定性分析的出峰顺序是:1.甲基汞;2.乙基汞。各组分与标准谱图相对照以保留时间定性。定量分析则通过色谱峰高或峰面积,在标准曲线上来查出各组分的浓度。水和沉积物的可检出浓度分别为0.01ng/L和0.02μg/kg。若样品中含硫有机物(硫醇、硫醚、噻酚等)均可被富集萃取,在分析过程中积存色谱柱内,使色谱柱分离效率下降,干扰甲基汞的测定。因此采用往色谱柱内注入二氯化汞苯饱和溶液,可以除去这些干扰,恢复色谱柱分析效率。[/size][/font][font=黑体][size=12pt]2.2 [/size][/font][font=黑体][size=12pt]高效液相色谱-原子荧光法[/size][/font][font=宋体][size=12pt] 方法取1L水样于分液漏斗中,加10g左右氯化钠,用40ml二氯甲烷分两次萃取,每次10 min。萃取液收集至50ml比色管中,加3ml反萃取溶液(1%半胱氨酸+0.8%乙酸铵溶液)进行萃取,振荡5min,吸取水层溶液进样。采用二氯甲烷萃取,半胱氨酸-乙酸铵溶液反萃取后,用高效液相色谱-原子荧光串连检测水质中烷基汞,色谱柱用 Venusil MP-18,150mm×4.6mm,5 m或与之等效的色谱柱,流动相为5%乙腈+0.462%乙酸铵+0.12%半胱氨酸。方法检出限随仪器灵敏度及样品基质不同而各异。甲基汞和乙基汞的检出限分别为0.3ng/L和0.6ng/L。 按相同的分析条件,进行纯水空白、校准曲线和实际样品的同步测定。甲基汞和乙基汞的标准色谱图,如图4所示。[/size][/font][font=宋体][size=12pt][img=,613,304]http://ng1.17img.cn/bbsfiles/images/2014/07/201407160012_506823_1634717_3.jpg[/img][/size][/font][font=宋体][size=12pt] 载流中用到的盐酸一般都含有无机汞,当无机汞含量高时,峰形拖尾会影响到甲基汞的色谱峰,一次须采用优级纯或纯度较高的盐酸来消除干扰。[/size][/font][font=黑体][size=12pt]2.3[/size][/font][font=黑体][size=12pt]存在问题[/size][/font][font=宋体][size=12pt] (1)在《地表水环境质量标准》(GB3838-2002)规定饮用水源地水中甲基汞不得超过1×10[sup]-6[/sup]mg/L,该标准规定的方法检出限不得大于1×10[sup]-8[/sup]mg/L。而GB3838-2002引用的[/size][/font][i][font=宋体][size=12pt]GB/T17132-1997[/size][/font][/i][font=宋体][size=12pt]环境烷基汞的测定[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法规定[/size][/font][font=宋体][size=12pt]的方法检出限刚好为1×10[sup]-8[/sup]mg/L,方法检出限基本满足,而沉积物的检出限则为2×10[sup]-8[/sup]mg/L。如果采用[/size][/font][font=宋体][size=12pt]高效液相色谱-原子荧光法,其[/size][/font][font=宋体][size=12pt]甲基汞的方法检出限则高达[/size][/font][font=宋体][size=12pt]3[/size][/font][font=宋体][size=12pt]×10[sup]-7[/sup]mg/L,显然已不能满足检出限应低于国家地表水Ⅰ类标准限值1/4的要求。[/size][/font][font=宋体][size=12pt]在《城镇污水处理厂污染物排放标准》(GB18918-2002)和《危险废物鉴别标准 浸出毒性鉴别》(GB 5085.3-2007)中规定烷基汞均不得检出,而标准引用的[/size][/font][i][font=宋体][size=12pt]GB/T[/size][/font][/i][font=宋体][size=12pt] [i]14204-93[/i][/size][/font][font=宋体][size=12pt]水质烷基汞的测定[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法[/size][/font][font=宋体][size=12pt]检出限同为1×10[sup]-8[/sup]mg/L。[/size][/font][font=宋体][size=12pt] (2)测试中样品处理是烷基汞析中最重要的步骤,分离技术的灵敏度尚不能满足微量有机汞的形态分析需求。由于全为手工分析,并采用自制巯基棉、吸附和萃取装置,人工操作的强度大和有机溶剂的暴露操作而不环保,因此方法的操作繁琐及回收率不稳定,有的实际样品甲基汞回收率低达67.5%。而采用高效液相色谱-原子荧光法虽较[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]法减轻了分析操作强度,但又带来检出限不满足的风险。[/size][/font][font=宋体][size=12pt] (3)对分析测定的影响因素较多,在检出与未检出之间受到试剂空白的影响较大。比如采用市售的盐酸等化学试剂的质量很不稳定,按GB/T14204-93规定需要的QC样品不易获得;易受玻璃仪器(分液漏斗、试管)、管路、仪器材质[font=宋体]的影响和存在记忆效应等。[/font][/size][/font]
一、事由常有《元旦献词》,这次我写个《新年献题》,向网友请教。现在存在一种说法,“现在的论文,不少是浮云”。其根据是,媒体报道,学术成果转成有效成果仅为10%。按此说法,10篇论文仅有一篇是有价值的新分析方法。现在存在另一种说法,“现在的论文,不是分析化学论文作者的分析化学的基本功好、不是作者学术水平高,而在于先进仪器卓越的性能:灵敏度、分辨率以及软件能力。”甚至有人将先进分析仪器称为傻瓜分析仪器,阿猫阿狗经短时间培训后都能操作……分析化学是否存在基本功问题?二、我想说分析化学当然存在基本功问题。我在南京大学化学系分析化学专业学习,深深地感到分析化学知识的广泛及深奥的科学道理。例如,很多分析化学问题的解决,与络合物原理有关;很多离子交换分离与离子的电荷及离子的半径密切有关……同时,分析化学实验/分析化学测试结果,与手上功夫极有关系,君不见:同样的分析方法,有的人做出的结果精度好,有的人做出的精度差。如同,患者到医院去抽静脉血,有的大夫稳准地插入血管,有的大夫插到血管边上,造成淤血。三、分析化学博士基本功试题1、有没有一个试题?如果谁解决了这个试题,能有效地平息他人对其学术水平的质疑。当然,这个试题应让质疑者不具备有解决这个问题的能力,即有点难度。举个例子:现在电视里播出的世界高水平运动员的跳高比赛,参加者至少有跳过2.20米的能力。如果A运动员有能力跳过2.20米,而你也作为一名跳高运动员,只能跳过2.00米,你还能质疑那位跳过2.20米的A运动员的能力吗?因为人家确实能比你可多跳20厘米。这20厘米,反应了跳高水平巨大的差距。2、分析化学博士基本功试题的要求:A、对试题的要求,既然是博士(青年)试题,当然要有一点难度。试题公布后能获大家认可。B、为避免“是由于仪器的卓越性能”的指责,试题应该尽可能不用仪器。C、答题者的水平高低,应尽可能在短时间内知晓,且为大家认可。D、样品来源随手可得。E、成功答题者会稍有成就感,如同你跳过了2.20米。3、分析化学博士基本功的试题(青年型)A、试题类型:容量法中的酸碱滴定。B、在100毫升烧杯中,加入25.0毫升1.60mol/L HNO3(即10%浓硝酸),加入50-100毫克铁丝,放置30分钟后,测定此时HNO3浓度。测定前可将尚未完全溶解的铁丝用塑料勺取出。若有不溶物,请过滤。C、假设:实验前后体积的变化为零。D、提示:你建立的方法,若加入回收试验虽很好,但很可能并不能证明方法准确。4、请您也提出试题,用您的智慧网友的智慧远超大家想像,我这试题真的是抛砖引玉。欢迎网友也提出试题,百花齐放,真正活跃学术气氛。您的试题,我一定认真去答题。5、今天是圣诞节,祝圣诞快乐!你领导又该给你们发年终奖了,祝这次的红包特别大!
市购的可溶性淀粉中约含20%水溶性的直链淀粉和80%的非水溶性的支链淀粉。前者与游离碘作用呈现深纯蓝色,后者则与游离点反应呈现弱紫色。因此,滴定终点在蓝色中带紫红色,会影响滴定终点的准确判别,滴定含硫小于0.01%的试样,可用红薯粉-盐酸作为吸收液。淀粉吸收液最好每滴定一次更换一次,这样可以保持条件一致,使得测定结果稳定。







 我要推广仪器
我要推广仪器
 下载APP
下载APP