我看到文献上写“用流动相稀释至刻度”,我要做土壤吸附,溶液都是用0.01M的CaCl2作溶剂的,在做标取时,我是用流动相稀释,还是用CaCl2溶液呢?流动相:乙腈:磷酸二氢铵=65:35.用流动相稀释,就是按照这个比例配成溶液,再用来稀释么?
请问各位,用流动相稀释是什么意思?比如我用的是安捷伦1200的液相色谱,标品浓度为1mg/ml,那么怎样设置,用流动相稀释成:0.005ug/ml,0.01ug/ml,0.05ug/ml……呢?

对于高效液相色谱分析样品的溶解与流动相的选择至关重要,除了遵循一般的流动相的选择之外,我们应该还要注意:用流动相稀释样品而不用吸光度比流动相强的溶剂稀释。下面的色谱图还隐藏着一些话题,分析讨论还能发现什么问题呢?http://ng1.17img.cn/bbsfiles/images/2014/12/201412091439_526337_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412091439_526338_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412091440_526339_2960432_3.pnghttp://ng1.17img.cn/bbsfiles/images/2014/12/201412091440_526340_2960432_3.png
求高手指点,标液的溶剂是正己烷,流动相是乙腈,正己烷和乙腈不互溶,这种情况要怎么稀释标液呢?
[color=#444444]使用离子液体(水不溶)萃取样品溶液以后,由于其粘度大,用乙腈进行稀释,但是稀释以后的溶液经试验证明不能完全溶解于乙腈:水=50:50的流动相中,请问此时还能用该流动相进行色谱分析吗?[/color]
请问各位老师,配置标准溶液的中间溶液,能不能用流动相稀释?
最近做液相,遇到的问题比较多,请各位大虾指点迷津。我想请问一下,标准品溶解后,用30:70的流动相定容。后来摸索条件发现,18:82的比例最合适。那先前用那个比例流动相定容配的标准储备液还能用不?定容用的流动相比例与实际进样用的流动相比例不同,是否会对实验结果造成较大的误差?谢谢!
各位老师,请问下,[url=https://insevent.instrument.com.cn/t/Yp][color=#3333ff]液质[/color][/url]流动相中加甲酸或者氨水时,甲酸和氨水还需要稀释吗
原先的分析条件:正相色谱,示差检测器,硅胶柱,流动相100%正己烷,稀释试样的溶剂也是100%正己烷,现在发现试样中有部分物质不容,准备稀释试样的溶剂改成90%正己烷+10%乙醇,但流动相仍旧用100%正己烷(因为是单泵,用2种流动相比较麻烦),请问这样分析有问题吗?会不会产生有些物质原来在90%正己烷+10%乙醇的溶剂中已溶解了,到流动相中仍旧会析出的现象?
今天科室液相流动相突然冒黄烟,充满整个房间,刺鼻,昨晚仪器开了一夜,今天早上实验人员处理完数据关机之后不久发生了反应,不知道原因,流动相为20%丙酮十80%乙腈(由1%甲酸稀释),请大家分析一下原因,谢谢!
这两天做桔子中的多菌灵,要用到流动相离子对试剂(7毫升磷酸、1g癸烷磺酸钠、5ml三乙胺稀释至1000ml),发现一个问题,三次都是这样,每次检测完,运行关机程序时,流动相比例从10甲醇:90水到90甲醇:10水最后这时总提示 lost prime.这时湿灌注和干灌注系统都不没有压力。厂家维修工程师总是判断说这是单向阀堵塞,流动相不干净;取下来超声2分钟,正常。从昨天到今天三次都是这样,做一次拆一次机子,亚历山大,请问这是仪器问题还是流动相问题
每次用流动相直接稀释参考品成标准曲线,检测样品的IgG浓度,HPLC检测样品中IgG浓度的专属性验证如何做?
我先用乙腈/水提取,然后用agilent C18填料做成的小柱净化,取上清液离心,再取离心后的上清液用10mM醋酸铵稀释,这样用微孔滤膜过滤后能直接上液相吗?(流动相:甲醇/水=45/55),不需要再向样品中加入甲醇吗?我以前做的时候流动相是甲醇/水,样品一般用甲醇溶解啊。谢谢啊
各位大神,我看文献流动相是乙腈,样品是用乙腈溶解的,昨天做实验的时候我们师姐问我溶解样品的乙腈需要稀释多少倍?溶解样品的乙腈不是买回来的乙腈(色谱级别)吗?还需要稀释后才能溶解样品?用什么溶解呢?文献中只是说乙腈溶解样品后需要稀释成梯度,以便做标曲,没说乙腈在溶解前要稀释后才能使用啊,请问乙腈在使用前需要稀释吗?用什么稀释?稀释的标准是什么?
关于流动相的问题所选用的流动相是乙腈 而用的是之前配好的用甲醇稀释的标准溶液 对实验结果或者出峰情况影响大么?有哪些影响?谢谢大家了!
我的流动相组成是:40%的四丁基氢氧化铵3.25m L + 125mL甲醇——用水稀释到1L。我用0.45有机膜(尼龙66)过滤,刚开始根本下不了,后面产生了很多气泡,整个抽滤过程花了半个多钟,平时过滤纯甲醇之类是很快的。问题:我认为加了甲醇了就应该用有机膜过滤,但是这个过滤速度来看是不是我选膜不对啊,我该用什么滤膜呢?
请问各位,正相色谱法检测样品含量所用的样品稀释剂是否可以改变。比如,大豆油含量测定时样品稀释第一步用的是正已烷和异丙醇(1:1)混合溶液,第二步稀释是用流动相。我可以把第二步的流动相换成1:1的混合液吗?如果换了,对测定结果及色谱系统是否会有影响。各位如果没有检测过,可以从理论上帮助我分析一下吗?非常感谢!
使用水或流动相作为替代基质的时候,后续加入提取试剂,不是相当于又稀释了一些倍数吗,这样回收率岂不是很低?比如A纯溶液:10μl质控样品+190μl水+内标B先前处理:100μl水+200μl甲醇混匀,取190μl+10μl质控样品+内标C正常:10μl质控样品+190μl水+内标+200μl甲醇这样起码C溶液中目标化合物相比B溶液中目标化合物的浓度又稀释了2倍,这样回收率会偏低吧?
研究表明,当流动相的A0.7的时候,基线噪声会显著增加,一般选择的吸收值0.5。当A1.0时,基本就不能用使了。对于示差折光检测器,主要考虑样品和流动相的折光率,这里不 再赘述。选用的溶剂粘度要低、沸点适中使用低粘度溶剂,可减小容质的传质阻力,降低柱压,利于提高柱效。从分离制备和色质连用考虑,沸点要适中,低沸点的溶剂有它的优点;但仅仅用 于分析,沸点太低,反而由于溶剂的挥发,造成保留时间的变化。尽量不用高毒性的溶剂不只是环境,单从我们自身的安全考虑,当然用低毒溶剂最好。溶剂对样品有足够的溶解力样品如果不能溶解在选用的溶剂里,还怎么分析?但如果样品在流动相里溶剂仍然不大,可以用样品的最佳溶剂先溶解,再用流动相稀释,就 可以了。知道了上面选择溶剂的一般原则,就不难理解为什么不同乙醇、丙醇做流动相的原因了,因 为它们的粘度大。 还有丙酮,虽然粘度和毒性较低、溶解度大、极性适中,但用于它的截至吸收波长为330nm, 所以不常用,如果样品的吸收大于330nm,其实用丙酮是一个不错的选择。
你觉得离子对流动相是哪一种更好:1、称取适量的离子对试剂,溶解,用酸调至一定的PH,再与有机相混合或混合后再调PH。如药典中的盐酸肾上腺素注射液,维生素B6等。2、称取适量的离子对试剂,溶解于有机相,再与已调至一定PH的缓冲溶液混合。如盐酸班布特罗片。盐酸班布特罗片:0.15%辛烷磺酸钠的甲醇溶液-乙腈-磷酸盐缓冲液(取磷酸二氢钾6.8g,加水适量使溶解,用磷酸调节pH值至3.0,加水稀释至1000ml)(34:11:55)为流动相;一个是没有另加缓冲溶液,一个是加入其它缓冲溶液,哪个更合理,不知道2不加其它缓冲盐会如何?
研究表明,当流动相的A0.7的时候,基线噪声会显著增加,一般选择的吸收值0.5。当A1.0时,基本就不能用使了。对于示差折光检测器,主要考虑样品和流动相的折光率,这里不 再赘述。选用的溶剂粘度要低、沸点适中使用低粘度溶剂,可减小容质的传质阻力,降低柱压,利于提高柱效。从分离制备和色质连用考虑,沸点要适中,低沸点的溶剂有它的优点;但仅仅用 于分析,沸点太低,反而由于溶剂的挥发,造成保留时间的变化。尽量不用高毒性的溶剂不只是环境,单从我们自身的安全考虑,当然用低毒溶剂最好。溶剂对样品有足够的溶解力样品如果不能溶解在选用的溶剂里,还怎么分析?但如果样品在流动相里溶剂仍然不大,可以用样品的最佳溶剂先溶解,再用流动相稀释,就 可以了。知道了上面选择溶剂的一般原则,就不难理解为什么不同乙醇、丙醇做流动相的原因了,因 为它们的粘度大。 还有丙酮,虽然粘度和毒性较低、溶解度大、极性适中,但用于它的截至吸收波长为330nm, 所以不常用,如果样品的吸收大于330nm,其实用丙酮是一个不错的选择。http://ng1.17img.cn/bbsfiles/images/2015/12/201512011315_575751_1858651_3.png
如果这样都不能岂不是流动相就选择错误了?没发生什么问题,看到版主回复多多的问题突然想到的。
各位大侠,跪求帮助。刚刚接触高压色谱仪。目前存在如下问题:流动相本底测试时,当流动相A切换成流动相B时,光电检测器就会有一个较大的峰。例如:流动相A切换为流动相B时,本底有一个向上的峰;流动相B切换成流动相A时,有个向下的峰。请问该峰的原因是什么呢??,有啥办法能够消除该峰。
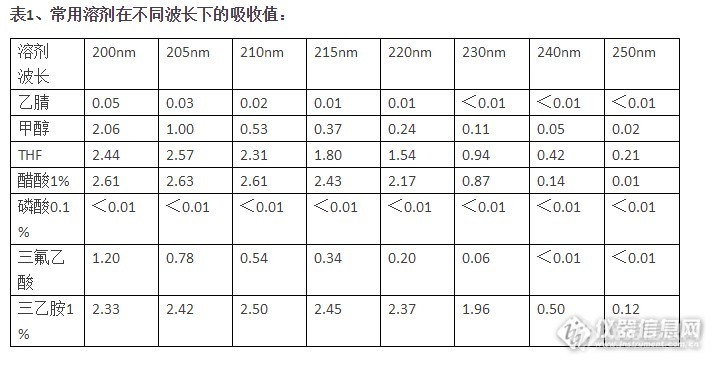
一、HPLC流动相溶剂选择的一般原则: 1. 溶剂要与使用的检测器匹配对UV-Vis,主要考虑本底吸收,下面是常用溶剂在不同波长下的吸收值: 200 205 210 215 220 230 240 250 乙腈 0.05 0.03 0.02 0.01 0.01 0.7的时候,基线噪声会显著增加,一般选择的吸收值1.0时,基本就不能用使了。对于示差折光检测器,主要考虑样品和流动相的折光率,这里不再赘述。 2. 选用的溶剂粘度要低、沸点适中使用低粘度溶剂,可减小容质的传质阻力,降低柱压,利于提高柱效。从分离制备和色质连用考虑,沸点要适中,低沸点的溶剂有它的优点;但仅仅用于分析,沸点太低,反而由于溶剂的挥发,造成保留时间的变化。 3. 尽量不用高毒性的溶剂不只是环境,单从我们自身的安全考虑,当然用低毒溶剂最好。 4. 溶剂对样品有足够的溶解力样品如果不能溶解在选用的溶剂里,还怎么分析?不屑多解释。但如果样品在流动相里溶剂仍然不大,可以用样品的最佳溶剂先溶解,再用流动相稀释,就可以了。知道了上面选择溶剂的一般原则,就不难理解为什么不同乙醇、丙醇做流动相的原因了,因为它们的粘度大。还有丙酮,虽然粘度和毒性较低、溶解度大、极性适中,但用于它的截至吸收波长为330nm,所以不常用,如果样品的吸收大于330nm,其实用丙酮是一个不错的选择。
在做药典中“头孢呋辛”的测试时,药典规定选择下面的流动相,--------pH3.4 醋酸盐缓冲液(取醋酸钠0.68g,冰醋酸5.8g, 加水稀释成1000ml,用冰醋酸调节pH 值至3.4)-乙睛(85∶15)为什么这么选择呢?
[color=#444444]我要测的样品是水溶液,可以用流动相(乙腈和水)稀释样品后再进样吗?跟直接进水溶液效果一样吗?测出来的量会不会有差别?谢谢了![/color]
试验目的:为该品种后期研究如有关物质和含量测定摸索流动相。试验条件:【含量测定】照高效液相色谱法(中国药典2010年版二部附录V D)测定。色谱条件与系统适用性试验用十八烷基硅烷键合硅胶 为填充剂;取醋酸铵3.96g,加水720ml使溶解,加乙腈280ml、三乙胺10ml,用冰醋酸调节pH值至5.5为流动相;检测波长为295nm。分别取盐酸帕罗西汀、去氟帕罗两汀与N-甲基帕罗西汀对照品各5mg,置同一10ml量瓶中,加流动相 溶解并稀释至刻度,摇匀,作为系统适用性试验溶液,取20μl注人液相色谱仪,记录色谱图,出峰顺序为去氟帕罗西汀峰、盐酸帕罗西汀峰、N-甲基帕罗西汀峰,理论板数按盐酸帕罗西汀峰计算不低于3000,盐酸帕罗西汀峰、去氟帕罗西汀峰与 N-甲基帕罗西汀峰之间的分离度均应符合要求。测定法 取本品10片,精密称定,研细,精密称取适量(约相当于帕罗西汀10mg),用流动相溶解并稀释制成每 lml中约含0.lmg的溶液,摇匀,滤过,精密量取续滤液20μl注人液相色谱仪,记录色谱图;另取盐酸帕罗西汀对照品,同法测定,按外标法以峰面积计算,即得。色谱柱信息:序列号(SN):W11212195。典型色谱图:http://ng1.17img.cn/bbsfiles/images/2013/02/201302051437_424683_1621890_3.gif对照品和样品色谱峰比较:http://ng1.17img.cn/bbsfiles/images/2013/02/201302051501_424684_1621890_3.gif试验总结:采用中国药典2010年版二部收载的方法,进行原料药的含量标定,其系统适用性结果符合要求,拖尾因子和分离度不是很理想,在后期研究准备调节流动相成分比例,以期得到改善。

今天做实验的时候,发现流动相中杂质竟如此之多呀,大家请看图片!!!http://simg.instrument.com.cn/bbs/images/brow/em53.gifhttp://ng1.17img.cn/bbsfiles/images/2016/01/201601151324_581957_1987954_3.png条件为辛烷磺酸钠1.8 g,无水硫酸钠20.0g,加磷酸盐缓冲溶液(0.2mol/L的磷酸二氢钾用0.2mol/L的磷酸调节pH=3.0)50mL,加乙腈50ml,用水稀释至1000mL,过滤。这样的流动相,如果偷个懒不过滤或者过滤不好的话,对于色谱柱的伤害真是不容小觑呀!!!都是一些常用试剂,为什么会如此脏???原因正在排查中……http://simg.instrument.com.cn/bbs/images/default/emyc1010.gif
最近买了几个威灵顿的标准品,溶剂是甲苯。而我用的液相流动相是乙腈/水,采用梯度洗脱(初始比例是40/60)。看了一些关于溶剂效应的经验帖子和文献之后,打算使用乙腈对购买的标品进行稀释配制成储备液,然后再用乙腈/水(40/60)的比例逐级稀释配制标准曲线。最后上机测定。现在我不能确定我的想法是否可行,由于标品比较贵,不敢轻易的尝试,希望得到高人指点!感激不尽!
降低稀释的强度(如增加水相比例、调整pH等)或更换稀释剂为流动相,2、在保证检测灵敏度的前提下,降低进样量。3、增加峰形前延抑制器,峰形前延抑制器的设计原理是,在色谱柱前端引入一个大小合适的空腔,使样品到达色谱前被存留于空腔内的流动相(色谱柱平衡时流动相会充满空腔)稀释,使“样品溶剂”经稀释后更接近流动相的组成,实现“样品溶剂与流动相的组成接近”,并取代“用流动相溶解样品”的解决方案。







 我要推广仪器
我要推广仪器
 下载APP
下载APP