从低角度衍射数据和图谱中都可以得到哪些有用的信息?现在做孔材料的人也挺多的,这些材料在低角度处会出现衍射峰,以前来我们这测样的就是想看看在他们期待的位置是否有峰出现,可最近我听说根据出现的峰的位置和峰形能够得到好多信息,比如孔的大小,孔率等等,具体我也不知道都有哪些,希望做这方面研究的人士能够在此详细谈谈,期待中...........
请问哪位大侠?我要表征纳米钨粉,可以用小角度衍射与小角度散射是测什么?以及两都有何区别?测试标准如何定?谢谢!!!!!!!1
TEM刚刚开始做,有两个问题请教各位老师:1. 我在看老师做TEM的时候,发现他在未加选区光阑的情况下按SA Diff按钮,本来应该出现菊池线,但是我却看到出现很整齐的衍射斑,就像加了选区光阑一下,这是为什么呢?是否是样品太薄?2. 之前做衍射的时候都只记录相机常数(CCD相机,虽然标定的时候也用不着),今天发现老师还让记录倾转角度(TX and TY),不知道倾转角度是用来做什么?有什么用途?先谢谢了!
标定过程中d值误差一般在1%左右,那么衍射角度的误差一般在多少?另外我的试样属于快速凝固,是不是误差范围可以放大一些,一般为多少?衍射角度差了0.6度,是不是差的太多了,那位高手指点一下,感激不尽!不知道黄老师在不在?
请老师指教小角度X光衍射测量粉体粒度的原理及方法!!!
用X射线小角度衍射作出来的图怎么跟pdf卡片对照阿?

求助大家,探测器卡在了低角度,手动调好之后发现标样测试衍射峰向右偏移2°多,然后用玻璃狭缝对光,操作按照图中所写,铜吸收片不知道是插在光管这端还是探测器那端,两边都试了试,reference and offset determination调整了两次,结果越调误差越大,后来在-1到1的区间都测不到峰了,求助大家,有几个地方实在不懂,铜吸收片到底是插在光管还是探测器一端?现在衍射峰偏移角度达2°,具体应该怎么调回来?下面的对光步骤哪里有问题吗,之前看工程师来调试的时候不是Rocking扫描模式,而是Theta模式,但是问工程师一直没回复[img=,690,548]https://ng1.17img.cn/bbsfiles/images/2021/07/202107101845059657_7874_4071974_3.jpg!w690x548.jpg[/img]
XRD仪器,型号为[font='Times New Roman']D/MAX2500VL/PC能不能做小角度(0-10度)衍射?谢谢![/font]
请教各位掠入射衍射小角度X射线有什么区别?那个对于分析纳米薄膜的晶体结构更有利?希望大家能多给介绍一下他们的功能。谢谢啦!
单位有台理学 X射线衍射仪(D/max2200VPC),用的很少,觉得太浪费了。最近在猛学相关知识。理学 X射线衍射仪(D/max2200VPC)能不能作小角度衍射?如果能,还请各位具体介绍一下如何操作。
请问:XRD谱图分析中,如何确定衍射角度才科学合理?谢谢
我使用的是FEI F20电镜,在每次装入样品后,都需要调节样品的共心高度。在获得共心高度前,调节束斑,发现束斑汇聚到一定程度时,在荧光屏上可观察到衍射花样,一般为多晶环和衍射斑点,而此时并未在衍射模式下,这是为什么?按照工程师的说法,通过调节z轴,使花样消失,样品就处于共心高度,这是为什么?还有我为什么通过SAED得到的衍射花样没有上面的漂亮,为什么不一样?百思不得其解?求教各位!
请教高手.测量外延薄膜时,低角衍射和高角衍射怎么测?用的是philips衍射仪.
[font=黑体, SimHei][size=16px]点击链接查看更多:[url]https://www.woyaoce.cn/service/info-14048.html[/url]射线衍射仪技术(X-ray diffraction,XRD)。通过对材料进行X射线衍射,分析其衍射图谱,获得材料的成分、材料内部原子或分子的结构或形态等信息的研究手段。X射线衍射分析法是研究物质的物相和晶体结构的主要方法。当某物质(晶体或非晶体)进行衍射分析时,该物质被X射线照射产生不同程度的衍射现象,物质组成、晶型、分子内成键方式、分子的构型、构象等决定该物质产生特有的衍射图谱。X射线衍射方法具有不损伤样品、无污染、快捷、测量精度高、能得到有关晶体完整性的大量信息等优点。因此,X射线衍射分析法作为材料结构和成分分析的一种现代科学方法,已逐步在各学科研究和生产中广泛应用。[/size][/font][font=黑体, SimHei][size=16px]测试内容[/size][/font][font=黑体, SimHei][size=16px]1. D8 Advance X射线衍射谱中的衍射峰与晶体中的不同晶面为一一对应关系,可以标定出各个衍射峰对应的晶面指数。根据衍射峰的位置、衍射峰的强度和形状,通过索引已建立的XRD标准卡片库,可检索出样品中存在何种物相。 [/size][/font][font=黑体, SimHei][size=16px]2. 物相定性分析 结晶度及非晶相含量分析 结构精修及解析 物相定量分析 点阵参数精确测量 无标样定量分析 微观应变分析 晶粒尺寸分析 原位分析 残余应力 低角度介孔材料测量 织构及ODF分析 薄膜掠入射 薄膜反射率测量 小角散射[/size][/font][font=黑体, SimHei][size=16px]可检测范围[/size][/font][font=黑体, SimHei][size=16px]1.常用于无机物。[/size][/font][font=黑体, SimHei][size=16px]2.有机晶体单晶不合适。[/size][/font][font=黑体, SimHei][size=16px]3.角度5-80度,快扫,慢扫。[/size][/font][font=黑体, SimHei][size=16px]4.物相是定量分析,相对定量。[/size][/font][font=黑体, SimHei][size=16px][/size][/font]
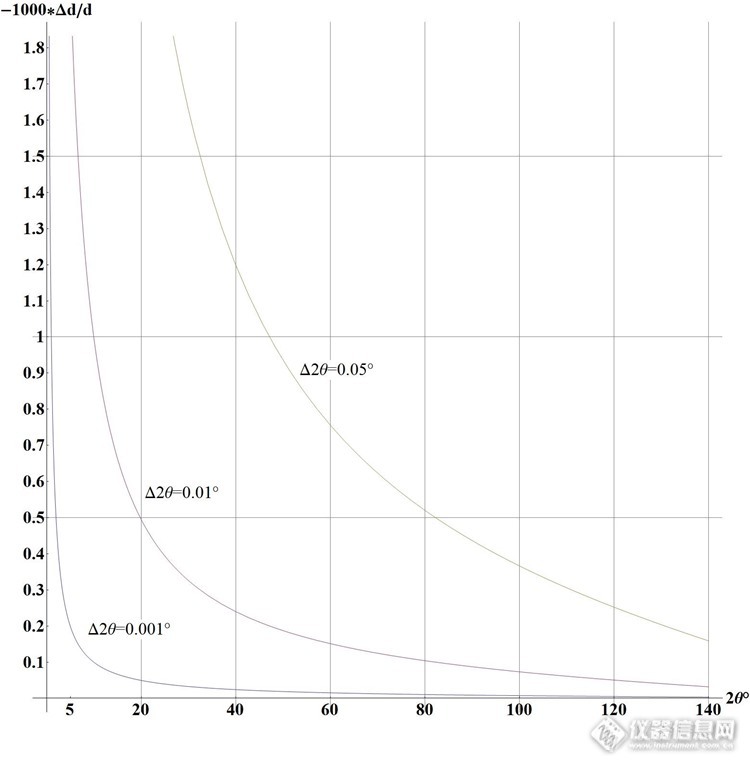
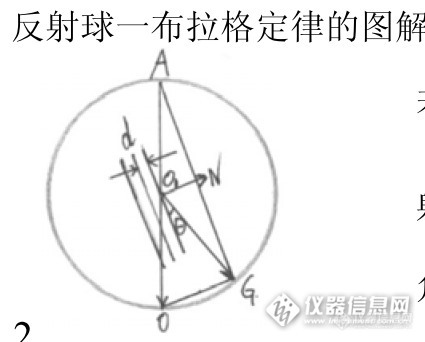
怎样用好你的衍射仪 ---- XRD的基础知识Xiaodong (Tony) Wang, Curtin University不久前有版友问起要XRD的基础知识, 说实话这应该去踏踏实实的看一本介绍XRD的书. 版友们虽然可针对某具体问题回复, 但不太可能具有系统性. 但为了避免版上只有几个高手过招的局面, 我还是应该给新手们做点普及基础知识的工作. 虽然主要是写给XRD衍射仪使用者的, 但租仪器的用户看了也会有帮助, 因为你付了钱, 就要对实验结果有要求.要用好XRD衍射仪, 先要明确你想要从衍射实验中得到什么; 你能调整哪些变量; 怎样调整这些变量以获取你想要的结果; 此外还要定期检查衍射仪的性能.一般来说我们从粉晶XRD谱中能得到的结果包括:1.鉴定物相 — 那么你需要准确的衍射强度以及产生这些衍射的准确的晶面间距2.定量相分析 — 那么你需要可重复的衍射强度(强度的统计性要好, 磨细你的样品10微米)3.解晶体结构 — 那么你需要可重复的衍射峰形+峰越多越好(角度分辨率要好以防止峰重叠, 尽量测到高角)4.求晶粒尺寸(相干衍射畴尺寸) — 那么你需要尽量减小仪器峰宽以突显样品展宽5.求应力和应变 — 同上, 尽量测到高角6.求晶胞参数 — 那么你需要准确的峰位(晶面间距) (要分清是固溶体还是化合物)但实际上: 峰位能测到多准: IUCr曾经做过Round Robin世界上多个实验室的衍射仪测Znic Oxide和Calcite的峰位, 其误差能达到0.05 2θ°强度能测到多准: 世界上多个实验室的衍射仪测Znic Oxide和Calcite的强度误差分别能达到13%和50% (由于后者还有择优取向问题)此外还要考虑你的样品是什么样品:如果是多聚物, 小晶粒如催化剂: 它们的衍射峰可能很差, 需要入射X射线强度大, 还可能是馒头峰所以角度分辨率其实不重要, 注意馒头峰太宽可能和背景无法区分开了.如果是半导体, 蛭石, 药物: 它们的结晶一般很好(峰尖锐), 所以角度分辨率很重要. 注意因为机械或化学处理它们常常出现多形 (将改变晶体学对称性)如果是多相无机混合物: 则应该考虑各相的峰位是否重叠: 如果没有重叠则应尽量增大入射强度, 追求高信噪比以尽量获取痕量物相. 如果峰位有重叠则要追求好的角度分辨率和好的信噪比以尽量区分主相.对扫描速度是否有要求(你付得起多少个小时做一个样): 可根据其调整入射强度, 步长, 计数时间, 和狭缝宽度等你拿到一张XRD衍射谱, 它一般都包含以下的组成部分:粉晶XRD衍射谱实际上是"衍射强度"对"2倍衍射角"的散点图. 所谓的"衍射强度"其实包含X射线衍射信号, X射线散射信号, 和X射线荧光信号. X射线衍射信号中还有你不需要的衍射波长(Kα波长以外的其他波长)引起的信号, 需要剔除 (当然现在的全谱拟合是把Kβ也拟合进去).X射线散射信号中分样品相干散射和非相干散射, 这些会构成谱图的背景. 此外还可能来自样品架的散射也是需要避免的, 它会在低角处产生额外的高背景.X射线荧光信号需要避免, 它会造成均匀(随2θ不变)背景. 因为入射光子的能量是一定的, 荧光产率上去了, 衍射信号就少了, 记住你是在做XRD不是做XRF.目前的精修软件一般可拟合衍射信号, 但是背景里面的散射和荧光是不可拟合的(也很难区分开), 所以做实验(扫描)的时候就要尽量减小这些非衍射现象. 典型的例子: 不能用Cu靶XRD扫铁矿样品, 因为会产生荧光, 要换成用Co靶XRD来做.光源有哪些:实验室X射线光管(X射线的产生装置)基本上是靠高能电子束轰击阳极金属表面而产生X射线. 不同的阳极金属会给出不同能量(波长)的X射线. 常见的阳极靶材有: Cu Co Cr Fe Mo等 显然在参见的测试范围内(5-140 2θ°), 这些波长所能测试的d值范围是不同的, 如下图所示: http://ng1.17img.cn/bbsfiles/images/2014/08/201408310221_512277_1986542_3.jpg这个图的作用是让你正确选择起始角度, 曾经有版友问粘土矿物的001峰怎么没有, 给出的谱图却是从10°开始扫的- -|||基本上扫XRD谱主要是扫晶面间距d值, d值能测到多准? 你对Bragg公式做全微分就能看出来: 主要是看测角仪的角度能测到多准. 如下图. (注意全微分后θ是弧度. 我看了很多书里这个图的y轴都是画错了的, 就是没注意到这个问题) http://ng1.17img.cn/bbsfiles/images/2014/08/201408310225_512278_1986542_3.jpg所以峰位测得准,你的d值才会准. y轴的负号表示:高估角度将会低估d值.那么为什么角度会有偏差呢? 主要是因为你的衍射实验可能有如下的主要偏差来源:1. 样品面高度不在仪器圆中心 (这是最常见的也是唯一可简单避免的偏差, 因为下面的偏差不容易发现与矫正)2. 光路各组件没对准 (工程师调的时候就要他对准)3. 两个转臂2:1角度关系不准 4. 2θ零点误差5. 低原子序数元素样品的穿透太深, 造成平均衍射面低于仪器圆中心6. 平板样品偏差7. 光线在测角仪轴方向上有发散, 再密的sollor狭缝也是有宽度的8. 计数器记录误差9. 由Kα2带来的峰形畸变你可控制的变量:1. 光源: 选择X射线光管阳极金属材料, 在常见的靶材 Mo, Cu, Co, Fe, Cr 的Kα能量中找出对你的样品中各物相的质量吸收系数小的, 且各相间的质量吸收系数相差不大的. 用点光源还是用线光源(目前普遍用的是线光源long fine focus). Kβ滤片根据阳极靶材的不用而不同. Mo靶用Zr滤片, Cu靶用Ni滤片, Co靶用Fe滤片, Fe靶用Mn滤片, Cr靶用V滤片. 或者考虑用不用, 用什么单色器, 是放在入射束还是反射束. 光管阳极发光区域射向各个方向的强度是不同: 基本上沿被轰击的矩形区域呈圆柱状分布: Be窗口只采用低出射角的光线主要是为了让出射X射线更细, 其实强度在垂直方向上确实最强的. (现在有人用光纤把射向其他方向X射线也利用起来.) 如果要用line-fine focus, 阳极靶上受轰击面积将非常小, 尽管光管功率开低, 减少受照射面积使能量密度W/mm^2升高的. 当面积持续减小使能量密度高于350W/mm^2以上时, 普通水冷已经不够散热了, 需要改用转靶或者液态金属阳极.光管出射的X光线既包含连续谱也包含特征谱, 但你的XRD实验只需要一种波长的X射线, 这就需要滤光: 用对应的kβ滤光片可以可以去掉大部分kβ和连续谱. 探测器本身也有一定的能量分辨率, 闪烁计数器最宽(差), 正比计数器稍微好点, 反射束单色器更窄, 最窄的还是入射束单色器. 但要注意: 越窄也意味着XRD谱图的强度更低, 你需要在强度和角度分辨率中作权衡. 总的来说, 优良的单色器可以减小半高宽, 使峰之间的重叠减小.2. 样品: 虽然大多数粉晶XRD是用的Bragg-Brentano反射几何, 但你也可以把样品装进毛细管中用Debye-Scherrer透射几何的, 粒径一定要细10微米, 如果是针状或者板状的样品要防止择优取向, 要用侧装样品架.3. 狭缝: 一般是固定宽度狭缝, 也有程控狭缝随2θ逐渐张开的, 入射狭缝和接受狭缝的宽度都决定强度和角度分辨率.4. 信号检测: 扫描程序由样品的类型而定, 主要是决定扫描角度范围, 步长, 每步驻留时间. 峰尖锐的把扫描步长设密点, 峰宽的把步长减小,适当增加驻留时间.衍射仪各光路组件对最终峰形的影响:1. 光源: 光管内钨灯丝发射电子轰击到阳极靶上是基本呈矩形(长l近似于钨灯丝长度,宽w),在常用的long-fine focus 下,从光管出来的x光线宽度为w Sinα,长近似于l. 光源峰形基本上是对称的Voigt峰形. 2. 降低轴偏差的soller狭缝: 用于防止光线偏出仪器圆所在平面: 如光源左端的光线射到样品再射入接受狭缝右端将使被记录信号的2θ偏低. 注意在高角下此误差将使峰位往高角移动: 这是因为光源左端的光线射到样品右端再射入接受狭缝的左端.3. 平板样品偏差: 大家会用AutoCAD的去画个图可知, 只要不是射向仪器圆心的光线, 都会被样品反射到接受狭缝的低角处. 所以平板样品偏差将使峰形往低角偏.4. 样品穿透等效于样品平面低于仪器圆中心, 将使峰形往低角偏.5. 发散狭缝和接收狭缝再窄也有宽度, 这会使峰形增宽.6. 各组件若对得不好也将产生一个对称的发散.所以, 除了kα2和高角轴偏差会使峰位往高角移以外, 其他都使峰位往低角移.发射狭缝有固定的也有程控的. 固定
样品为线状,相用SAED确定生长方向。发现得到衍射花样后拍图相时随放大倍数样品变化角度。是不是不改变相机长度直接拍的那个图是正确的?TEM为JEM 1011实验室另一台好用的2011坏了,只能凑合用这个破东西了。用2011时好像没发现这个情况。大家给出个注意吧。
本人初涉入TEM,对标注衍射斑还不很清楚,求高人指点。最近拍出的衍射斑(物质是面心立方)和标准衍射斑对照,,r1/r2以及角度都相同,就是方向不同,旋转一个角度刚好相同了。可以旋转吗?旋转之后可不可以对着标准衍射斑直接标注?我感觉晶带轴相同,旋转不影响,不知道对不对啊?求大神指点
[size=4]我是做插层膨润土复合吸附剂的[/size][size=4]改性后的膨润土做出来的x衍射图 部分的首峰是向低角度移动 那么根据布拉格公式 它的层间距就会变大有些的首峰发生了漫射 消失了 是不就可以认为它的层间距大于2nm了 不知道群里有没有人做插层复合材料的 对于剥离型插层的话 它的衍射峰首峰会消失 那么衍射峰首峰消失的材料我是不是就可以认为它就是剥离型插层了[/size]
[size=4][font=楷体_GB2312]X射线衍射法因晶体的是单晶还是多晶分为x射线单晶衍射法和X射线多晶衍射法。 [b]单晶X射线衍射分析的基本方法[/b]为劳埃法、周转晶体法和四圆单晶衍射仪法。书上还会有别的方法,因不太常用在此不再啰述。现在最常用的是四圆单晶衍射仪测单晶。 [b]劳埃法[/b]改变波长、以光源发出连续X射线照射置于样品台上静止的单晶体样品,用平板底片记录产生的衍射线。根据底片位置的不同,劳埃法可以分为透射劳埃法和背射劳埃法。背射劳埃法不受样品厚度和吸收的限制,是常用的方法。劳埃法的衍射花样由若干劳埃斑组成,每一个劳埃斑相应于晶面的1~n级反射,各劳埃斑的分布构成一条晶带曲线。 [b]周转晶体法[/b]:周转晶体法以单色X射线照射转动的单晶样品,用以样品转动轴为轴线的圆柱形底片记录产生的衍射线,在底片上形成分立的衍射斑。这样的衍射花样容易准确测定晶体的衍射方向和衍射强度,适用于未知晶体的结构分析。周转晶体法很容易分析对称性较低的晶体(如正交、单斜、三斜等晶系晶体)。 [b]四圆单晶衍射仪法[/b]是转动晶体。以四个圆的转动变量φ、χ、ω和2θ进行晶体和计数器的转动,以实现倒格点与埃瓦尔德(Ewald)衍射球球面相遇产生衍射的必要条件。φ圆对应于安置晶体的测角头的自转转动,χ圆对应于测角头在其所坐落的仪器金属χ环内侧圆上的转动,ω圆对应于金属χ环绕中垂线(Z轴)进行的转动,2θ圆则对应于为保持衍射方向相对于入射X射线为2θ的角度所需进行计数器的转动。是常用的测量单晶衍射的方法[/font][/size]
用X射线照射DNA分子,观察射线在照相底片上产生的点子(衍射花样),计算点子的分散角度等(每一点子的分散角度代表DNA分子的一个原子的位置或若干原子团的位置)推测分子排列。 最关键的第51号图谱是下图,1952年5月拍摄。 照片中心X射线反射(使X射线底片变黑)的图象是交叉的,说明它是螺旋形的,顶部和底部最浓黑的部分,说明嘌呤碱和嘧啶碱垂直于螺旋轴,每隔3.4埃规律出现一对。 对A型DNA、B型DNA拍了好多张X射线衍射图谱,这两张是截面的,也有丝状(链形态的),可以得到34埃的数据。富兰克林还发现在翻转180度之后看起来还是一样,沃森与克里克在得到这一信息后,意识到两条链是反向的。 在得到51号图时,还得到的了一些数据。 1953年2月24日富兰克林经过计算分析得出双股螺旋的结论,而沃森与克里克则是尝试以双螺旋模型与这些数据信息吻合。当时自然杂志同时发表了三篇论文,另二篇是威尔金斯的和富兰克林与蓝道夫的。 解读DNA晶体X射线衍射图谱,要用到很复杂的数学计算。 X射线衍射原理: 1912年劳埃等人根据理论预见,并用实验证实了X射线与晶体相遇时能发生衍射现象,证明了X射线具有电磁波的性质,成为X射线衍射学的第一个里程碑。当一束单色X射线入射到晶体时,由于晶体是由原子规则排列成的晶胞组成,这些规则排列的原子间距离与入射X射线波长有相同数量级,故由不同原子散射的X射线相互干涉,在某些特殊方向上产生强X射线衍射,衍射线在空间分布的方位和强度,与晶体结构密切相关。这就是X射线衍射的基本原理 。
埃瓦尔德球上面表示的衍射只是代表了一个晶面,而且球的方向是以入射电子方向作为竖直方向,而晶面对应的晶带轴与竖直方向是成θ角的。可是选区电子衍射不是以晶带轴作为竖直方向,然后电子束从各个角度入射,从而得出该晶体晶带轴对应的所有晶面衍射斑点吗?想请教下大家,选区电子衍射的所有斑点是怎么来的呢,是电子束从各个方向入射得到的吗?样品放置应该是晶带轴朝竖直方向放置吗,不是埃瓦尔德球的方式放置的吧?

衍射花样的形成 在EBSD测试中,扫描电子显微镜(SEM)电子束入射到倾斜70°度的晶体样品表面。(图 2)EBSD衍射花样获得的基本原理非常复杂,这里简要介绍了它的一些主要的工作原理。入射电子束与样品表面原子碰撞发生弹性散射,伴随着一小部分能量损失,从而形成了一个电子发散源。而其中一些电子于原子面入射角度满足布拉格方程http://www.ebsd.cn/images/articles/70/image002.gif(公式1)其中,n是整数,λ是电子波长,d是衍射面间距,θ是电子入射方向与衍射面之间的夹角。这些电子发生衍射形成了与每一个衍射面相对应的一系列成对出现的大角度锥面。在荧光屏上进行成像时,这些经过信号放大的锥面间电子强度区域形成了电子背散射衍射花样—菊池带花样(Kikuchi band)。http://ng1.17img.cn/bbsfiles/images/2012/07/201207311531_380891_1622447_3.jpg
有一个事情一直让我犯难:因为X射线衍射实在有点专业性质,向一般人解释不清楚,那么如何向新生或一个外行参观者(经常性的一些什么长,主任,书记之类的人)介绍X射线衍射仪和它的功能呢?就我们的衍射仪功能来说,有这么一些:1 物相鉴定与物相定量2 微观应变与晶粒度测量3 残余应力测量,样品最大1公斤4 结晶度、石墨化度测量5 结构精修与晶胞参数测量6 高温原位衍射7 小角度衍射8 极图,反极图和ODF测量请大家积极发言,用简短的、能听懂的表达介绍衍射仪及其主要功能。要求:1 时间不超过3分钟。2 介绍衍射仪。3 介绍一点主要功能。4 可举例。5 最主要的,一个外行能听懂。分数只会给最佳的回复。
已经得到晶带轴为【111】方向的衍射图,怎么在这个基础上得到[112]方向的衍射图呢,请各位大佬指导一下[img]https://simg.instrument.com.cn/bbs/images/default/em09511.gif[/img]
我现在做的工作是将活性组分负载在载体上,请问使用X射线衍射可不可以测定负载的活性组分的颗粒大小,X衍射小角度衍射分析可以吗?
布鲁克D8 Advance光路调零后,仪器自带标准样品35.149,测量为35.150,但测量硅粉的角度异常,28.443的测量值为28.445,88.032的测量值为88.002,高角度偏低很大,不知什么原因,请高手赐教!谢谢

[img=,690,453]http://ng1.17img.cn/bbsfiles/images/2017/08/201708042059_01_3162679_3.jpg[/img]已知成分为 Fe ,S, 一张衍射图的长短轴之比大约为R1/R2=2.670,角度约为86或94,记得刚开始标定透射衍射图片是查卡片,可以根据成分和长短比来确定物相和指数,但很久没做了,现在手头找不到数据,请高手帮忙标定下可能值。
对于未知物相的标定,我知道大致有三种方法。一个就是查询astm卡片。第二个就是,利用已知的一些参数(甚至是假设的)来自己计算一个数据表,通过这个数据表可以查询相应的hkl以及d值的对应关系。第三种就是利用双倾台在不同旋转角度下拍摄3张以上的衍射照片。 大致的方法我知道些,但是具体的操作我还是不太明白。比如,我们现在做的是一个全新的物相,当然不可能在astm卡里面查到了,所以第一种方法不行。由于我们生长的晶体尺寸的限度,同步辐射也只能给出几个猜测的晶格常数的结果。我想是否可以利用这些结果自己制作一个数据表。以便验证同步辐射的结果。至于这个数据表怎么进行计算呢?~ 就是利用正空间a,b,c和beta(单斜)求出倒空间的a`,b`,c`和beta`么然后,再带入几个hlk的值么?,但是我发现在astm卡里001,和002所对应的d值不是2倍的关系啊。这又是怎么回事呢!? 至于第3种方法。曾经看过郭可信在物理学报1982,7月。vol.31,No.7的一篇文章。每太看明白,[em49] 。但是感觉总体思想是首先得到一幅衍射图样,然后,保持一列衍射斑点不动,倾动样品,使得与其他列衍射斑点的距离发生变化,得到很多组衍射花样。这里面有很多问题啊,怎么保证有一列衍射斑点不动?只倾动一个方向,那能找到正带轴么?还有样品台倾动的方向怎么能保证和这列衍射斑点垂直呢,难道要调整磁转角? 刚接触电子显微镜几个月,也就是基本操作。但是一直对衍射标定很迷惑。多谢大家不吝赐教~。。。
特征X射线及其衍射 X射线是一种波长很短(约为20~0.06┱)的电磁波,能穿透一定厚度的物质,并能使荧光物质发光、照相乳胶感光、气体电离。在用高能电子束轰击金属“靶”材产生X射线,它具有与靶中元素相对应的特定波长,称为特征(或标识)X射线。如铜靶材对应的X射线的波长大约为1.5406埃。考虑到X射线的波长和晶体内部原子面间的距离相近,1912年德国物理学家劳厄(M.von Laue)提出一个重要的科学预见:晶体可以作为X射线的空间衍射光栅,即当一束 X射线通过晶体时将发生衍射,衍射波叠加的结果使射线的强度在某些方向上加强,在其他方向上减弱。分析在照相底片上得到的衍射花样,便可确定晶体结构。这一预见随即为实验所验证。1913年英国物理学家布拉格父子(W.H.Bragg,W.L.Bragg)在劳厄发现的基础上,不仅成功地测定了NaCl、KCl等的晶体结构,并提出了作为晶体衍射基础的著名公式──布拉格方程: 2d sinθ=nλ式中λ为X射线的波长,n为任何正整数。 当X射线以掠角θ(入射角的余角)入射到某一点阵晶格间距为d的晶面上时(图1),在符合上式的条件下,将在反射方向上得到因叠加而加强的衍射线。布拉格方程简洁直观地表达了衍射所必须满足的条件。当 X射线波长λ已知时(选用固定波长的特征X射线),采用细粉末或细粒多晶体的线状样品,可从一堆任意取向的晶体中,从每一θ角符合布拉格方程条件的反射面得到反射,测出θ后,利用布拉格方程即可确定点阵晶面间距、晶胞大小和类型 根据衍射线的强度,还可进一步确定晶胞内原子的排布。这便是X射线结构分析中的粉末法或德拜-谢乐(Debye—Scherrer)法(图2a)的理论基础。而在测定单晶取向的劳厄法中(图2b)所用单晶样品保持固定不变动(即θ不变),以辐射束的波长作为变量来保证晶体中一切晶面都满足布拉格方程的条件,故选用连续X射线束。如果利用结构已知的晶体,则在测定出衍射线的方向θ后,便可计算X射线的波长,从而判定产生特征X射线的元素。这便是X射线谱术,可用于分析金属和合金的成分。 X射线衍射在金属学中的应用 X射线衍射现象发现后,很快被用于研究金属和合金的晶体结构,出现了许多具有重大意义的结果。如韦斯特格伦(A.Westgren)(1922年)证明α、β和δ铁都是立方结构,β-Fe并不是一种新相 而铁中的α─→γ转变实质上是由体心立方晶体转变为面心立方晶体,从而最终否定了β-Fe硬化理论。随后,在用X射线测定众多金属和合金的晶体结构的同时,在相图测定以及在固态相变和范性形变研究等领域中均取得了丰硕的成果。如对超点阵结构的发现,推动了对合金中有序无序转变的研究,对马氏体相变晶体学的测定,确定了马氏体和奥氏体的取向关系;对铝铜合金脱溶的研究等等。目前 X射线衍射(包括散射)已经成为研究晶体物质和某些非晶态物质微观结构的有效方法。在金属中的主要应用有以下方面: 物相分析 是 X射线衍射在金属中用得最多的方面,分定性分析和定量分析。前者把对材料测得的点阵平面间距及衍射强度与标准物相的衍射数据相比较,确定材料中存在的物相;后者则根据衍射花样的强度,确定材料中各相的含量。在研究性能和各相含量的关系和检查材料的成分配比及随后的处理规程是否合理等方面都得到广泛应用。 精密测定点阵参数 常用于相图的固态溶解度曲线的测定。溶解度的变化往往引起点阵常数的变化;当达到溶解限后,溶质的继续增加引起新相的析出,不再引起点阵常数的变化。这个转折点即为溶解限。另外点阵常数的精密测定可得到单位晶胞原子数,从而确定固溶体类型;还可以计算出密度、膨胀系数等有用的物理常数。 取向分析 包括测定单晶取向和多晶的结构(见择优取向)。测定硅钢片的取向就是一例。另外,为研究金属的范性形变过程,如孪生、滑移、滑移面的转动等,也与取向的测定有关。 晶粒(嵌镶块)大小和微观应力的测定 由衍射花样的形状和强度可计算晶粒和微应力的大小。在形变和热处理过程中这两者有明显变化,它直接影响材料的性能。 宏观应力的测定 宏观残留应力的方向和大小,直接影响机器零件的使用寿命。利用测量点阵平面在不同方向上的间距的变化,可计算出残留应力的大小和方向。 对晶体结构不完整性的研究 包括对层错、位错、原子静态或动态地偏离平衡位置,短程有序,原子偏聚等方面的研究(见晶体缺陷)。 合金相变 包括脱溶、有序无序转变、母相新相的晶体学关系,等等。 结构分析 对新发现的合金相进行测定,确定点阵类型、点阵参数、对称性、原子位置等晶体学数据。 液态金属和非晶态金属 研究非晶态金属和液态金属结构,如测定近程序参量、配位数等。 特殊状态下的分析 在高温、低温和瞬时的动态分析。 此外,小角度散射用于研究电子浓度不均匀区的形状和大小,X射线形貌术用于研究近完整晶体中的缺陷如位错线等,也得到了重视。 X射线分析的新发展 金属X射线分析由于设备和技术的普及已逐步变成金属研究和材料测试的常规方法。早期多用照相法,这种方法费时较长,强度测量的精确度低。50年代初问世的计数器衍射仪法具有快速、强度测量准确,并可配备计算机控制等优点,已经得到广泛的应用。但使用单色器的照相法在微量样品和探索未知新相的分析中仍有自己的特色。从70年代以来,随着高强度X射线源(包括超高强度的旋转阳极X射线发生器、电子同步加速辐射,高压脉冲X射线源)和高灵敏度探测器的出现以及电子计算机分析的应用,使金属 X射线学获得新的推动力。这些新技术的结合,不仅大大加快分析速度,提高精度,而且可以进行瞬时的动态观察以及对更为微弱或精细效应的研究。 爱心捐助
版面很多网友由于刚接触TEM的衍射花样,所以有一些基础问题觉得需要这里讲一下,有什么错误大家也请帮忙指出,多谢! 我这里就谈一下简单衍射花样的标定,所谓简单,就是各个晶系里面的单晶衍射花样,没有缺陷,没有超结构,没有厚样品造成的高阶劳埃带,只是物质的纯相造成的衍射花样。有了这个基础,理解了一些,往下才能做的扎实。1. 一般的物质衍射花样都是已知的物质,顶多也就是已知的几种里面的一个。所以在确定哪几个物种之后,去找一下相关物质的PDF卡片,网上有一个软件PCPDFWIN,可以方便查讯电子版的PDF卡,下载位置,看看这个帖子,提到了下载的具体目录:http://www.instrument.com.cn/bbs/shtml/20060418/398715/ 2. 找到了相应的PDF卡,那么就是要测量衍射花样了。衍射花样的拍摄要严格按照操作规程来,尤其要注意在拍摄时样品聚焦尽量准确。另外,无论底片拍摄还是CCD拍摄,一定要保证用标准样品做了校正。3. 接下来就是测量衍射点对应的d值。对于底片来说就是测量衍射点到中心透射斑的实际距离R,然后根据d = R/(L×电子波长),其中L是相机常数,底片上写着,单位是cm,电子波长一般的电镜书上都有,200 kV电镜是0.00251 nm。代入计算即可得到相应的d值。选取两个相邻且最靠近中心斑点的衍射点,二衍射斑点以夹角接近或者等于90度为好。选取测量d值之后,二者同中心斑点连线的夹角也要测量一下。对于CCD相机拍摄的衍射花样,对应的都有标尺,d值测量就是量取衍射点到透射斑的距离后取倒数即可。角度测量可以通过量取衍射点到中心斑连线对应control对话框的R值(角度),二者相减即得。4. 将计算的d值和PDF卡相对应,看最接近哪个面的数值,querida说过,这个测量会有一定的误差,有相近值时,需要通过夹角来确定。方法是,选取两个比较可能的面,然后代入相应晶系对应的公式,计算夹角,如果和测量值很接近,就算是找对了。Ustb版主说过,计算值和测量值应该相差很小,0.1-0.2度的范围。 至于计算两个面夹角的公式,可以去找郭可信先生写的那本《电子衍射图在晶体学中的应用》,Page104-105上有具体的公式,其中的hkl值都是你要计算的面对应的值,abc是你确定晶相的晶胞参数,PDF卡上都有,r1*r2*分别指的是两个面的d值倒数。5. 确定了两个方向的衍射点,那么接下来就是确定投射方向,也就是面的法线方向是什么带轴,这个querida朋友已经写了,我这里引用一下:“FFT后的一个斑点对应这正空间一族晶面,这一族晶面和这个斑点的矢量方向垂直,当一张图片上任意不在同一直线上的2个斑点知道后,那么入射电子束也就是带轴的方向就知道了,具体可用h1 | k1 l1 h1 k1 | l1h2 | k2 l2 h2 k2 | l2u v wu=k1l2-l1k2v=l1h2-h1l2w=h1k2-k1h2你可以适当化简达到互质的3个数。”这里的uvw就是法线方向,一般用[uvw]表示。6. 另外高分辨透射电镜有时候观察纳米粒子由于条件限制未必能得到好的衍射花样,这个时候高分辨图像的二维晶格像也能做类似的标定,以CCD相机拍摄的数码照为例,可以有两种方法:第一种:a. 选取两个相邻晶面,量取d值,注意尽量取多一些晶面层,这样量的误差较小,还要提醒一下新手,量的时候是取晶面层之间的垂直距离,而不是两个亮点之间的连线距离(除非是矩形或者正方形)。并量取二面的夹角b. 对应PDF卡,大致确认晶面,夹角计算见第4步。c. 同4中的部分,确认投射方向第二种:a. 对二维晶格像按alt键选取正方区域做FFT,得到类衍射花样。这个时候的衍射点就和拍摄的晶格像对应,量取衍射点和中心斑的距离同第3步。b. 其余的标定同上。****注意:凡CCD相机拍摄照片的处理软件均为DigitalMicrograph







 我要推广仪器
我要推广仪器
 下载APP
下载APP