文献解读丨基于LC-MS/MS结合蛋白沉淀和杂质提取技术的奥曲肽临床药代动力学研究
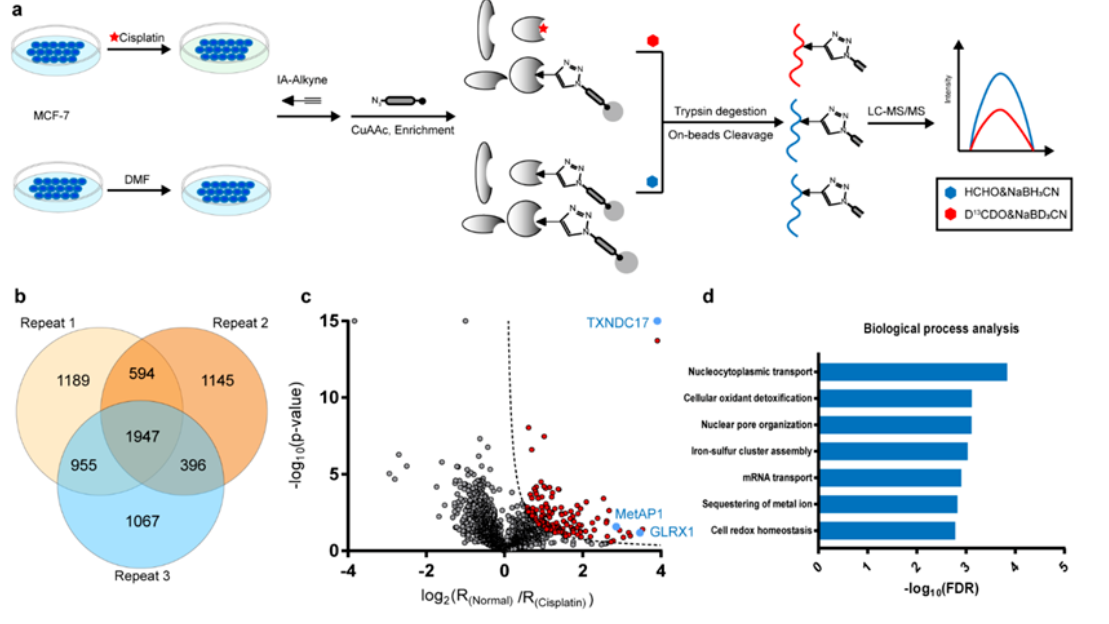
本文由中国药科大学天然药物国家重点实验室药物代谢与药代动力学重点实验室所作,发表于Bioanalysis (2015) 7(7), 885–894。 目的:建立生物样品中治疗性肽的定量分析方法。材料与方法:以奥曲肽为模型治疗肽,合成氧化奥曲肽作为内标。采用蛋白沉淀结合液-液萃取技术,有效提高了回收率,减少了内源性干扰。根据FDA指南,对血浆中奥曲肽的LC-MS/MS定量方法进行了优化和验证。结果:线性、选择性、准确度、精密度、稳定性、基质效应和回收率均符合FDA指南生物分析方法验证验收标准。该方法已成功地应用于奥曲肽的研究。结论:该方法可用于奥曲肽和其他治疗性肽的药代动力学研究。 图 大鼠血浆中一种奥曲肽样品制备过程 图 奥曲肽和氧化奥曲肽的代表性质谱图。(A)乙腈蛋白沉淀法制备的空白血浆 (B)蛋白沉淀结合液液萃取法制备的空白血浆 (C)加入奥曲肽(50.0 ng/ml)和内标的空白血浆 (D) 加入奥曲肽(0.5 ng/ml)和内标的空白血浆 (E)大鼠静脉注射奥曲肽0.1 mg/kg后2h采集血浆。AQT: 奥曲肽 C-A: 氧化奥曲肽图 大鼠血浆中奥曲肽的校准曲线,浓度范围为0.5-500.0ng/ml表 大鼠奥曲肽静脉注射0.1 mg/kg和灌胃45 mg/kg后的主要药代动力学参数 在临床研究中,已经发表了几种用于奥曲肽定量的生物分析方法。然而,由于这些文献在样品处理技术、内标选择和检测方式等方面的差异,很难选择一种稳定的检测方法。此外,就我们所知,对奥曲肽的临床前药代动力学特征知之甚少。 • 开发的以氧化奥曲肽为内标的大鼠血浆中奥曲肽的LC-MS/MS定量方法已根据美国指南进行了充分验证。• 线性、准确度、精密度、基质效应和回收率符合FDA指南中的生物分析方法验证验收标准。• 所建立的LC-MS/MS方法在大鼠血浆中表现出优异的性能。• 上述方法已成功应用于奥曲肽的药代动力学研究。• 目前奥曲肽在大鼠体内的药代动力学研究将为奥曲肽的开发和合理使用提供有益的信息。• 目前开发的方法只需稍加修饰,可以用于其他治疗性肽类的药代动力学研究。 文献题目《PK study of octreotide based on LC-MS/MS combining protein precipitation and impurity extraction technique》 使用仪器岛津LCMS-8050 作者Qian Wang**,1, Yan Liang*,1,Tai Rao**,1, Lin Xie1. Wei Ye1, Hanxu Fu1, Liiun Zhou1,Rong Xing1,2, Yuhao Shao1,Jingcheng Xiao1, Dian Kang1&Guangji Wang11 Key Lab of Drug Metabolism&Pharmacokinetics, State Key Laboratory of Natural Medicines, China Pharmaceutical University, Tongjiaxiang 24, Nanjing 210009, China2 Department of Pharmacy, the First Affiliated Hospital of Bengbu Medical College, Bengbu, Anhui, 233004,China* Author for correspondenceTel.:+862583271128FaX:+862583302827liangyan0679 @hotmail. com**Authors contributed equally






















 我要推广仪器
我要推广仪器
 下载APP
下载APP