各位大虾, 请问XPS元素定量分析准吗?我做的主要是O和Si(〉10%),其中掺杂有少量的P(《1%), 原来一直是由XPS来定量分析成分的。但是我现在怀疑其测量含量O/Si的准确性。请教各位有什么看法。

我的样品是金刚石单晶颗粒,合成时加入了硼,尺寸0.3-0.6mm的都有,想对金刚石中的硼元素做定量分析,不知用什么仪器合适,特请教。谢谢。[img]http://ng1.17img.cn/bbsfiles/images/2005/11/200511300935_11068_1195592_3.jpg[/img]
新手,有以下问题请教:1. 就目前EDS硬件而言,SEM-EDS可否检测0.3at%的Cu并精确定量分析?oxford和Edax哪家仪器较好?2. 目前有哪些标样生产商,市场上可有为掺杂Cu的标样出售或定制?3. 目前国内在SEM-EDS轻元素及微量元素定量分析方面哪个单位较牛?望不吝赐教,万分感谢!
sem能做元素定性定量分析吗?做到什么程度?
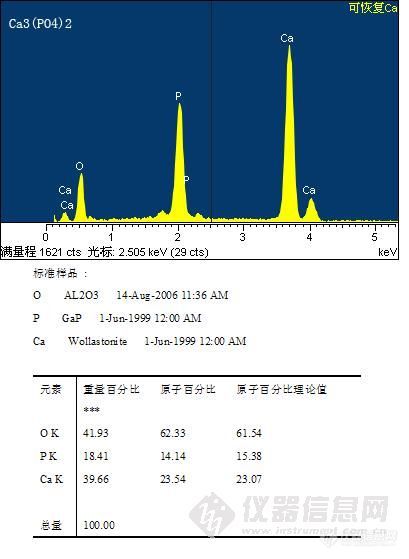
按照仪器要求做好最优化和标准化,超轻元素的定量分析有望得到比较理想的结果。(牛津能谱)http://ng1.17img.cn/bbsfiles/images/2010/11/201011081104_258043_1609375_3.jpg
粉末压片法测试岩矿定量和半定量分析都能分析那些元素最近,想搞 粉末压片法 不知道 都能测那些元素? 请高人指教
粉末压片法测试岩矿定量和半定量分析都能分析那些元素最近,想搞 粉末压片法 不知道 都能测那些元素? 请高人指教
直读光谱仪的元素定量分析,测量误差是否有标准要求?
今天第一次进入这个论坛,也发现了一些很有意义的帖子。我也对自己相对熟悉点的领域写了几个帖子,与大家分享。对任何分析仪器来说,定性问题容易解决,但遇到定量化问题,事情就变得异常复杂。EDS定量分析在目前也是个很有挑战性的问题,主要原因是目前EDS定量化理论就不完善,并且EDS定量化涉及到众多因素的影响。EDS的定量化目前主要还是以Cliff-Lorimer K因子为基础,但K因子不是常数,它和样品,电镜以及EDS探测器都有关系。样品包括样品成份,测量点的样品厚度等。电镜则包括加速电压,样品在电镜中的位置(主要是考虑减小x光的吸收以及减小二次荧光等)。EDS探测器的影响更大,其作用可以用探测器效率来表示,包括固体收集角(solid collection angle,角度越大则探测器效率越高),取出(take-off)角,窗口类型,窗口是否结霜等。当样品比较厚的时候,就应该考虑吸收校正以及荧光校正(数学处理比较复杂)。下面我简单介绍一下EDS定量分析的过程。(1)首先要有标样。标样就是和你要分析的样品有相近的成份的标准样。标准样的成份要已知,并且厚度要已知,一般可以从美国国家标准局或者国内的相关单位购买。然后测量标准样品的K因子。测量的时候要注意以下几点,(1)首先测量hole count,反应了在样品还没有被激发时电镜电子束流本身对EDS探测器计数的影响(2)对要测量成份的样品(一般是楔形样品),EDS探头要对着样品的薄区(减小特征x光逸出样品的路径,从而减小吸收)。(3)被测量元素的计数一般应在105以上,一般活时间live time要大于100秒。(4)计算K因子的时候考虑吸收校正。(5)多点测量。如果没有标样,那么利用确切已知成份的晶体也可,但必须知道厚度。(2)相同测量条件下测量自己的样品,得到元素的K峰的强度,经过吸收校正,利用已得到的标准样的K因子,得到自己样品的元素含量。一般EDS配备的软件给出的定量结果可以作为参考值来使用,因为其置信度不高,特别是对于超轻元素,B, C, N, O等,因为超轻元素的荧光产额低,并且吸收严重,所以EDS很难给出准确结果。最近有人参考电子探针的元素定量分析原理对Cliff-Lorimer因子做了改进,提出了一个新的因子,实际就是质量厚度。这种方法优点很多,测量时不需要预先知道样品厚度,可以同时测量出样品中元素的含量以及测量点的厚度(在测量点密度已知的情况下),并且这种方法对超轻元素的测量结果也相对准确,但这种方法最大的缺点在于,必须准确知道电子束流强度。EELS中的Faraday cup可以测量束流强度,所以这种方法对没有配备EELS的电镜是不适用的。有兴趣者可参考 M. Watanabe and D.B. Williams, J. Micro., Vol. 221, 2006, pp 89.今天想到哪就写到哪,可能会有一些遗漏或不太准确的地方。希望能对有兴趣的研究者提供一些有意义的信息。[em01]
做环境腐蚀试验,请问金属腐蚀产物中的氯元素定量分析用什么方法好呢?分析精度可以达到多少?
请问何处能用SIMS(secondary ion mass spectrometry) 定量分析微量元素,元素含量在ppm级。如能告知,不胜感激。有知道的朋友,麻烦给出测试单位详细的地址。再次感谢。
今天实验室来了两个X射线方面的专家,在期间我提到了压片法做矿石中的多元素的定量分析(高含量),专家的回答是,是实现不了的,因为矿物效应无法消除,所以无法准确测量矿石中的多元素。
纳米级的Co3O4,经过氮化处理后,想测一下表面有没有N元素,并进行定量分析,请问可以用那些测试方法?
钢铁企业是需要做夹杂物、偏析度、元素含量分布等定量分析的,不知有色行业是否也需进行这方面的定量分析?http://simg.instrument.com.cn/bbs/images/brow/em09511.gif
libs定量分析的标准曲线发,发哪个期刊合适
XPS与XRF的主要区别是什么?如果我要想进行固体粉末样品中各元素的定性和定量分析,我选哪一种分析手段比较合适呢?谢谢各位高手!
从原理上讲,原子发射光谱可以进行定性分析、半定量分析和定量分析。过去因为电弧光源和火花光源在性能上存在缺陷,使发射光谱在定性和半定量分析方面应用非常广泛,但其定量分析受到很大限制。ICP光源的出现彻底改变了这种状况,使发射光谱定量分析的应用越来越多,现已基本上成为元素分析的常规手段。但大家在用ICP进行定量分析的同时,还经常去做定性和半定量分析吗?
大家好,请问一下大家能介绍一下光谱分析的定量依据吗?我了解的光谱就是发射光谱和吸收光谱,吸收光谱就是[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url],发射光谱就是ICP,我觉得原子荧光也算发射吧?不知道原子荧光算那种还是新的名称? 我的理解是,吸收光谱分析的依据应该是朗伯-比尔定率,因为它就是一个入射光通过一个均匀介质(一定厚度的原子密度),利用吸收的光与[url=https://insevent.instrument.com.cn/t/Wp][color=#3333ff]原子吸收[/color][/url]成正比关系,符合朗伯-比尔定率。 那发射光谱的定量依据是什么?最近在网上看到说的是罗马金-赛伯公式,具体是:光谱定量分析依据试样中欲测元素的谱线强度来确定元素的浓度。元素的谱线强度I与该元素在试样中的浓度c的关系为:I=ACb,这个公式称为罗马金-赛伯公式,是光谱定量分析的基本公式。式中A及b是两个常数。常数A是与试样的蒸发、激发过程和试样组成等有关的一个参数;常数b称为自吸系数,它的数值与谱线的自吸收有关。所以只有控制在一定的条件下,在一定的待测元素含量的范围内,A和b才是常数。取对数得光谱定量分析的基本关系式lgI=blgC+lgA。 不知道我的理解是不是正确?不知道大家有什么想法或补充的,大家讨论讨论吧!!谢谢
请问:不知名样品怎么样对其中元素进行定性定量分析?广州或者深圳哪些机构可以做这方面的分析,多谢了.
都说能谱仪能半定量分析元素,能否比较准确的定量分析呢?下面是我们遇到的问题:一个未知样品,你全面扫描和部分扫描所分析出来的质量百分比是完全不一样的。
用EDS做定量分析的时候有两种方法,一种是standardless一种是有standard的,请问如果我要给一个未知样品做定量分析,我知道这个样品大概有哪些元素,而且肯定不止一种元素,但是我不知道他们之间的相对含量,如果给这样的样品建立standard。
求助:在EELS定量分析中,我选择软件中的Quantification和元素后,软件自动选择计算的范围,可不可以手动改变范围啊?
这两天看到有同学在讨论定量分析最优化,有感而发:定量分析最优化其实是一种无奈的选择,为什么说他是无奈的选择,一言蔽之:就是因为厂商采用了虚拟标样数据库作为定量参照系。相信用过的TX都知道,在采用虚拟数据库定量的系统里面,要得到非归一化结果,必须定量分析最优化,否则系统无法显示非归一结果。(因为结果会惨不忍睹,偏差太大)。这是虚拟标样定量(也叫半定量)最大的弊端。为什么虚拟标样定量法(不进行定量分析最优化)非归一化结果会很差呢?因为虚拟标样数据库都是在同一出厂条件下采集所得,当时采集的条件(如WD,HV,TOA等参数)和当前分析的条件完全不同,所谓差之毫厘,谬以千里,在分析条件都完全不同的情况下,把虚拟数据库作为标尺,无异于刻舟求剑,要想得到好的定量结果也是痴人说梦。定量分析最优化到底在优化什么?有说法是监测扫描电镜束流(相当于法拉第杯的作用),我的理解是这种说法欠妥。个人以为定量分析最优化通过采集标准样品如(Ti),通过采集的数据(里面包含了当前的分析条件)来比对虚拟数据库里面的数据,生成一个修正因子,通过这个修正因子来修正定量分析的结果。定量分析最优化的缺陷:1.修正因子是通过标准样品采集数据和虚拟数据对比所得,该因子只对所采用标样元素的修正有较好的表现,对于其他元素,特别是特征能量相差较大的元素修正表现并不好(这也是厂商要求用户尽量采用和所测样品特征能量接近的标样去做定量分析最优化的原因)。[color=#DC143C]注:不同元素(原子序数由小到大)在不同的分析条件下,受分析条件的影响因子是不相同的。通过经验公式可以使得影响因子有统计规则可循,但是误差相对比较大。[/color]2.分析条件改变,定量分析最优化必须重做文中观念并无权威出处,写出来只是给大家参考(上次写的东西有位老兄盯着我要出处,把我给盯傻了)呵呵,这次赶紧交代先。
无标半定量分析可以直接选定几个元素,建立方法,然后定性分析,得出结果可以用全扫描,把所有的元素选出,建立方法,定性分析,也会得到无标半定量的结果理论上,这两个数值,应该没有大的差异可是实际上,却相反想请教一下大家
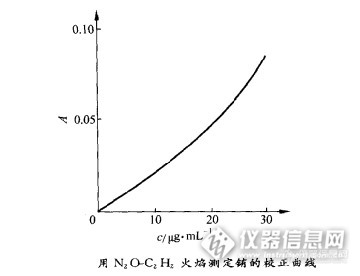
影响原子吸收光谱定量分析的因素原子吸收光谱定量分析涉及两个基本过程:①试样中被测元素转化为自由原子的化学过程;②蒸气相中自由原子对辐射吸收的物理过程。化学过程比物理过程更复杂,影响化学过程的因素比影响物理过程的因素更多。1 原子化过程的影响在推到原子吸收光谱定量分析的关系式A=Kc时,假定了一个基本条件:在确定的实验条件下,蒸气相中的原子数N与试样中被测元素的含量c成正比,N=βc,为此要求被测元素的原子化效率在确定的实验条件下是一定的。准所周知,在实际分析工作中所遇到的试样类型千变万化,即使是同一元素,在不同的试样内,由于基体特性各异和其他共存元素的相互影响,其原子化效率各有不同,有时甚至差别很大。原子化效率对实验条件非常敏感,在原子吸收这类高温动态测量中,实验条件的变动性导致原子化效率的改变,几乎是不可避免的。这是影响原子吸收光谱分析的准确度和精密度的主要因素。由此可以得出这样的结论,测定一种试样中某一元素的最佳条件,未必适用于另一种试样中同一元素的测定,必须针对具体分析对象,寻求某一元素测定的最佳条件。现在商品原子吸收光谱仪器中,厂家为用户所提供的预先储存在数据库内各元素的分析条件,多半都是用纯溶液样品得到的,只能作为选择实际分析样品分析条件的参考。计算机的广泛使用、原子吸收仪器自动控制系统的日益完善以及横向加热石墨炉和STPF技术的应用等,为获得稳定的原子化条件提供了可能性。化学过程是一个复杂的过程,有关影响化学过程的因素。2 辐射吸收过程的影响从光源的发射线考虑,在原子发射线中心频率V0的很窄的△V频率范围内,kv随频率的变化很小,可以近似地认为kv→k0,。当空心阴极灯光源的发射线远小于原子吸收线的宽度时,如下图所示,测得的吸光度可以近似地认为是峰值吸光度。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161143_573671_2352694_3.png随着空心阴极灯的灯电流增大,由于自吸和多普勒变宽效应增强,使光源发射变宽,对于低熔点金属Cd,Zn和Pb等元素空心阴极灯,光源发射线和原子吸收线宽度几乎达到同一数量级,使测得的峰值吸光度明显地降低,导致校正曲线严重弯曲。下图使用不同灯电流时所得到的镉校正曲线。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161144_573672_2352694_3.png在入射辐射中,若含有非吸收辐射,如连续背景辐射、空心阴极灯内稀有填充气体与支持材料以及其他杂质发射的辐射等,它们都可能出现在光谱通带内。当不存在非吸收辐射时,吸光度A=lgI0/I,当存在非吸收辐射i0时,吸光度A’=lg(I0+i0)/(I+i0),A’小于A0。i0在整个入射辐射中所占比例越大,A’比A小的越多。i0和I0比例一定时,I值越小,即吸收介质内分析原子浓度越高,i0的影响越大。非吸收辐射i0的存在,使测得的吸光度减小,校正曲线弯曲。从吸收谱线轮廓考虑,在通常的原子吸收光谱分析条件下,分析原子浓度都很低,共振变宽效应可以忽略不计。但是,当吸收介质的分析原子浓度高时,同种分析原子相互碰撞引起谱线共振变宽,使峰值吸光度减小。随着分析原子浓度增大,对峰值吸光度的影响增大,因此,造成校正曲线在高浓度区弯向浓度轴。这是导致校正曲线非线性化的重要因素。在建立峰值吸收的定量关系式http://ng1.17img.cn/bbsfiles/images/2015/11/201511161141_573669_2352694_3.png时,假定吸收谱线轮廓主要由多普勒变宽效应决定。事实上,吸收谱线轮廓不仅受多普勒变宽效应的影响,还与碰撞变宽,特别是洛伦茨变宽有关。在有些情况下,多普勒变宽与洛伦茨变宽是同一数量级,不能忽略其影响。洛伦茨变宽还引起吸收谱线轮廓的频移与非对称化,使得测定的吸光度不能代表峰值吸收,而是中心波长两侧的吸光度,其值低于峰值吸光度,导致校正曲线的非线性化。谱线的精细结构是影响吸光度测量的又一可能的因素。这些相差很小的谱线精细结构常常是简并的。对于很重和很轻的元素,其波长差超过了线宽,在这种情况下,测定的吸光度是精细结构内各组分的混合吸光度,而非单一纯组分的吸光度,故导致校正曲线的弯曲。当用锐线光源进行峰值吸收测量时,谱线的精细结构对吸光度测定的影响可以忽略不计。下表列出了某些元素共振线的同位素移值。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161145_573673_2352694_3.png从吸收介质内原子浓度考虑,在推到吸收关系式http://ng1.17img.cn/bbsfiles/images/2015/11/201511161142_573670_2352694_3.png时,认为入射辐射密度pv是不变的。很显然,只有在吸收层很薄或分析原子浓度很低时才是这样,这说明原子吸收光谱法主要用于痕量和超痕量元素分析。当被测元素的浓度高时,引起吸光度下降,校正曲线弯向浓度轴。由此可知,原子吸收光谱分析的校正曲线线性范围不会很宽,一般是1-2个数量级。在通常的原子吸收条件下,可以忽略激发态原子和元素电离的影响,但对于低电离电位元素,特别是在高温下,不能忽略电离对基态原子的影响。电离度随温度升高而增大,在一定温度下,随元素浓度增加而减小。元素电离的影响如下图所示,电离效应导致校正曲线弯向纵轴。http://ng1.17img.cn/bbsfiles/images/2015/11/201511161145_573674_2352694_3.png
我在做合金表面的XPS分析的时候没有考虑到Cu这种元素,结果老师做好后我在全谱中发现了这种元素,但没有对它进行分峰,所以也就没法进行定量分析,所以我想问一下哪位知道用全谱是否能进行定量分析,怎么做呢?谢谢了!
X射线荧光光谱法进行定量分析的根据是元素的荧光X射线强度Ii与试样中该元素的含量Ci成正比:Ii=Is×Ci式中Is为Ci=100%时,该元素的荧光X射线的强度。根据上式,可以采用标准曲线法、增量法、内标法等进行定量分析。但是这些方法都要使标准样品的组成与试样的组成尽可能相同或相似,否则试样的基体效应是指样品的基本化学组成和物理化学状态的变化对X射线荧光强度所造成的影响。化学组成的变化,会影响样品对一次X射线和X射线荧光的吸收,也会改变荧光增强效应。例如,在测定不锈钢中Fe和Ni等元素时,由于一次X射线的激发会产生Nika荧光X射线,Nika在样品中可能被Fe吸收,使Fe激发产生Feka。测定Ni时,因为Fe的吸收效应使结果偏低,测定Fe时,由于荧光增强效应使结果偏高。
[size=5][font=KaiTi_GB2312]各位,新手上路,请教一个问题。最近做了几个定量检测的样品,仪器做完第一个样品,并把结果算出来打印出来了。第二个样做了五六个小时,分析进程显示所有元素都分析完了,可是样品还没有拿出来,分析结果也仅有强度,没有含量。后来我们又编了几个定量分析的样,一起能顺利的做完。各位有遇到过这种情况吗?什么原因呀?都怎么解决的啊?谢谢啦![/font][/size]
[b]影响定量分析结果准确性的因素[/b]色谱定量分析中,每个操作步骤和每个色谱条件的选择都会对色谱定量分析结果的准确性产生影响,稍有不慎,就会使定量分析结果产生较大的误差,甚至会得到完全错误的结果。下面就影响色谱定量分析结果准确性的几个主要因素进行详细讨论。一、样品制备被分析的样品确定后,首先要把其中的欲测组分转化成能用色谱进行分析的实验用样品,这一过程称为样品制备。在样品的制备过程中,欲测组分不能发生任何损失。如要将欲测组分转变成另一便于色谱分析或检测的形态时,可将不能气化的欲测组分通过衍生化转变成可以气化的形态,以便于[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]的分析 也可将没有紫外吸收的欲侧组分通过衍生化转变成有紫外吸收的形态,以便于液相色谱的紫外检测器检侧等.这些转变一定要是定量的,最好转化率达到l00%(转化率达不到l00%时,一定要知道准确的转化率,以便最后计算欲测组分的含量).在选择提取或溶解欲侧组分的溶剂时,对于[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]色潜分析要考虑这一溶剂应能气化,气化温度要低于欲测组分的分解温度,气化后的气体不与色潜柱中的固定相发生化学反应 对于液相色谱分析要考虑这一溶剂应与液相色谱的洗脱液互溶,而且不与洗脱液和色谱住中的面定相发生化学反应。在用液相色谱分析时,最好选用所选择的洗脱液作为溶剂来溶解或提取被分析样品中的欲测组分,这样可以避免溶剂峰的于扰。在样品制备过程中,要同时考虑将被分析的样品中,可能下扰欲测组分定量的物质尽可能的分离出去。当欲测组分含量很低时,还要考虑通过样品制备使欲测组分在试验样品中的含量得以提高(即通过样品制备,将欲测组分加以富集)。以便于最后的色潜分析。 样品制备过程中,影响色谱定量分析结果准确性的七要因素是被分析样品中的欲测组分是否能100%定量地转入到制备好的,可用于色谱分析的实验用样品中去,这可用回收率试验来检验,即可用已知量的欲测组分,用样品制备的同样方法处理,将这一欲测组分制备成可用于色谱分析的实验用样品,再定量测定欲测组分的量。将这一侧定结果与原来取的已知量相比,即可得到这一样品制备方法的回收率。当祥品制备方法的回收率较低时,宜用标准加入法定量,这样可以补偿欲侧组分在祥品制备过程中的损失,使色谱定量分析结果更加准确、可靠。[color=red]最后有整篇文章的PDF文档,如果看了觉得有用的朋友可以下载。[/color]
[size=4][font=宋体] [/font][font=Times New Roman]X[/font][/size][size=4][font=宋体]射线荧光仪定量分析计算过程的基本步骤为:[/font]1. 标准曲线模型的建立;2. 在标准曲线模型设定的各元素测量条件下,测量样品,获得测量谱图;3. 谱处理过程,得到各元素的荧光强度;4. 基体效应校正过程,在标准曲线模型设定的校正方法计算样品中各元素含量; FP法基体效应校正,需要计算光管原级谱分布,可参考附件中的计算程序。下面详细介绍这四个部分。[font=宋体]关键词:XRF,模型,谱处理,基体效应,定量分析[/font][/size]







 我要推广仪器
我要推广仪器
 下载APP
下载APP