最近用顶空做溶残,同一份溶液等体积放到几个瓶里扎样,总是有随机不出峰的,换了新的顶空进样针也没用,并且新针进了6针拆下来发现针前端有点弯了(之前旧针取下来发现也会变弯),还可能是顶空进样器哪有问题吗?已用直接进样方式排除[url=https://insevent.instrument.com.cn/t/Mp]气相[/url]的问题。顶空型号:hp7694
版友问题,做乙醇甲苯残留溶剂顶空进样不出峰,做其他的直接进样正常,会有什么原因呢?
如题,做溶剂残留,进空白(就是进了一针空气)也出峰了,我的主要检测溶剂是乙酸乙酯,环己烷和正己烷,这三个物质都出峰了,我的色谱柱以前没做过这些实验,请问是不是顶空系统被污染了,因为我之前的进样浓度有点偏高。
各位高手,本人现在在做气相色谱的计量检定,我用得事SE-54柱子,进样口温度230,检测器温度230,柱温箱温度160℃,都是国标条件,可是为什么只出溶剂峰,不出正十六烷峰呢?后来还出了倒峰,这是怎么回事啊?同样地条件、柱子和标准物质,用安捷伦6890来检定,出峰情况挺好的,正十六烷峰在8min左右就出来了,现在检定国产气相,就是不出峰,希望各位高手帮忙分析下原因,指点下我,谢谢啊!万分感激!!!
以前很少做气相,现在要做溶剂残留,主要是测乙酸乙酯,仪器是岛津2014,顶空进样。有以下几个问题,求助各位,先谢谢啦1、溶剂应该选什么,我们目前是用二甲亚砜做溶剂,是否可以,二甲亚砜的沸点比较高,应该比乙酸乙酯出峰要晚吧2、大家有没有用内标,是不是自动进样就可以不用内标呢?3、用什么色谱柱合适,我用的是HP-5的柱子4、气相做样品时,每做完一针是否需要在初始温度平衡一段时间
我在做样品的乙醇残留,溶剂是DMSO,顶空进样,以前这个检验重复过多次,都能顺利进行,前几天,进样,标样峰诡异的减小,甚至没峰。我的对策:联系工程师,清洗vent阀,采用蒸汽洗脱程序,清洗顶空系统。第二天再次进水溶乙醇标样,出峰正常,再进DMSO溶标样,仍然峰很诡异的小,换了DMSO,重复操作,结果一样。求助高手,分析一下什么原因,哪里的故障?多谢!
电子元件研磨后的黑色粉末样品,内含金、银、铜,金、银含量在100ppm左右。测银含量,样品加1:1硝酸,电热板加热,过滤时会出现白色沉淀,加硝酸,沉淀溶解了。定容后又出现白色沉淀,咋回事呢?怎样消除之使溶液澄清?
作仪器分析时,样品处理时有沉淀出现,需要用滤纸过滤后才能进样,因为是定量分析,涉及到定容问题,请问是先定容再过滤进样,还是先过滤再定容进样。
首先可以肯定二氯甲烷、三氯甲烷、四氯化碳、二氯乙烯、三氯乙烯、四氯乙烯等几种卤代烃类是溶剂是不可以直接进1uL到ECD检测器里的。(一氯乙烯是气体)但是可以作为被检测的物质,先溶进其他溶剂里,再进仪器。可是有些东西,在ECD检测器上同样响应非常灵敏,却似乎是可以作为溶剂直接进入ECD检测器的?目前我已经遇到了的有二硫化碳。进样之后会在ECD上出一个很大的平头峰。还有哪些溶剂是会让ECD出平头峰但是可以进样的呢?(碎碎念:氯苯这类东西似乎很少有拿来做气相色谱用的溶剂的)
各位老师,配制标准溶液混标时,总体积超出定容体积,怎么配才正确?比如配混标10ppm,定容10ml,标液已超10ml,是要把全部标液吹至近干,再用溶剂定容10ml?还是直接把超出定容体积之外的标液吹至容量瓶刻度下,再用溶剂定容到刻度?哪种配制方法误差相对少点呢?或者是各位老师还有更准确的配制方法?
哪位老师按照国标5009做过顶空法测定溶剂残留,我这边顶空是半自动的,进标液都不出峰,直接进样不用顶空就可以出锋,请问该如何处理?
大家好,请问你们有没有做过有关溶剂残留(主要是甲酸残留)的实验?我现在用的是安捷伦6890的仪器+顶空进样器,之前我尝试顶空做乙酸的残留,对照溶液用的是乙酸+水,也用过乙酸+DMF,出峰都比较杂乱,响应值也很小,完全不能判定对照品的峰是哪一个,实验失败。请大家有空帮忙分析一下原因。这次又需要做甲酸残留,请高人指点指点,衷心感谢!
[font=&]各位大神,我遇到顶空残留的问题,顶空进空气和水样都会有残留峰出现,而且测水峰面积比测空气大3-4倍,出峰时间在3.9和6.5分钟左右,但是进氮气就没有峰出现,顶空针,六通阀,切换阀,GC进样口,检测器口都洗过了,柱子也截了老化了,测试水和空气还是一样的峰[/font][font=&]空白溶剂是水 测乙醇残留 [/font][font=&]条件:恒温炉 80 样品流路 90 传输线105 [/font][font=&] 进样口 200 柱子程序升温50-220 [/font]检测器250 进样口压力42KPa 总流量8.1ml 柱流量 0.46ml 分流比10[font=&]GC是顶空和液体混用 液体进样测试的样品出峰面积有1亿多面积,也有1千多万的面积 [/font][font=&]最有疑问的是进了氮气就没有峰出现 [/font][font=&]现在怀疑分流流路和吹扫流路有残留[/font]且实验条件的流量偏小 但是测氮气也是一样的条件就没有峰就又矛盾了[font=&][/font][font=&] [/font][font=&] [/font]
顶空气相色谱法测定缬沙坦原料药中残留溶剂,以二甲基乙酰胺(DMA)作溶剂时,单独的对照不出峰,混合对照出峰,急求各位分析这是为什么?如何改进?顶空条件:DANI HSS86.50 顶空进样器,平衡温度100度,进样系统温度120度,传递管路温度120度,加压时间30秒,压力平衡时间5秒,注入时间30秒气相色谱条件:进样口温度200,检测器(FID)温度250,柱温:初始温度40,维持2分钟,以3度/秒升至100,再以30度/秒升温至200,维持2分钟;分流比10:1色谱柱:rtx-5ms,30m,0.25,0.251、以同样的样品(DMA溶解)手动进样出峰,但顶空不出峰。重复前期品种的方法,配置以水溶剂的甲醇、乙酸乙酯混合对照溶液,顶空进样,结果出峰,这是不是说明问题与DMA有关?2、DMA沸点166度,有没有可能是因为它在系统中冷凝?个人将顶空进样系统温度升至180度后,开始几针样品好了(但仍不确定是不是因为温度的原因),但过了一会单独的对照又不出峰了?3、因为中国药典标准中是用二甲基乙酰胺,所以用它了请各位帮忙分析一下,急死了!有什么没有说明白的地方,请指出,非常非常感谢!
溶劑有丙酮,丁酮,異丙醇等,它們是混合在一起的,溶劑含量較高。儀器:安捷倫6890和安捷倫7890、檢測器:FID想用現在的條件對揮發有機溶劑進行定量,需要知道方法和樣品前處理的方法,定量的方法用內標還是外標好?
岛津2030配HS-10的顶空进样器,装了一个多月,平时用自动进样,今天第一次用顶空。用乙腈定位,发现居然没有出峰!色谱柱是常用的溶剂残留用柱,SH-RTX-WAX 30*0.53*0.25的方法如下:炉温80流路100传输120恒温时间30分钟温度曲线:60℃平衡4分钟,4℃每分钟上升到100℃,再45℃每分钟上升到200℃,平衡4分钟方法也很普通,但是就是没有峰。第一次进了一个混合样,用DMF溶解的,出了一堆很小的峰,最大的也就8000uv第二次进了乙腈,没溶解。没出峰。图如下乙腈沸点80多℃,不应该没出来。请问可能是什么原因,那里出了问题?[img]https://ng1.17img.cn/bbsfiles/images/2019/10/201910101352349977_9635_3116636_3.png[/img]
定容液是否会影响出峰时间?怎样选择定容液和流动相?
最近,要组建一个小规模的新实验室,需要重新购置一整套实验室分析仪器,其中就要购买溶出仪。由于资金有限,不考虑进口的,要在国产里面选择。之前也接触过一两家国产溶出仪的产品,想分享我的使用心得,也想大家谈谈自己都使用什么国产溶出仪,效果如何,给我一个采购的参考。() 现在我工作的地方,有3台溶出仪。 其中1台是ZRS-4,原天津大学无线电厂的,应该来说是老字号了。虽说没有什么定时开机、关机等功能,但最基本的控温、转速功能,已经可以满足日常需求了。关键是性能稳定,少说也有十几二十年的历史了,现在还在一线工作,^_^,只是有一段很长时间没有,齿形带老化了,换过一套之外,主要零部件都完好无损。可能唯一的缺点,就是发热量有点大,如果不开空调,开机一段时间,机盖会热得烫手。所以一般不开空调的话,都打开机盖,好散热。曾经做释放度,连续3天满负荷工作,中途关机休息大概1个小时,都能挨过来,出乎我的意料。 另外2台是今年4月份买的,“天津HYD”生产的。才刚买不久就接二连三的出现问题。一个是转桨、转篮等出现锈迹现象,其中一根转桨更加表面爆裂,有黑色锈斑渗出,很是吓人。后来经协商,重新换了经防腐材料包裹的桨篮。虽说我们是做缓控释制剂,释放度一做就是24小时,介质一般是0.1M盐酸,但也不至于这么不耐用,严重怀疑桨篮的不锈钢材质是不是偷工减料,以低级别代替高级别材质,很失望。另外是 转杆转动时声响很大,齿形带磨损严重,后来厂家工程师到场检修更换了,才有好转。除了这个,还有就是电路板故障,两台都出现这种现象,电路板负责控制转速的插头烧糊了,经检查是接触不良造成过热,金属插头很单薄,仅靠一片金属片与金属柱接粗,后来直接将插头焊接在电路板上,就没事了。可见,还是材料没下足,可能是因为价格压得太低,材料就纷纷缩水了。 还有,就是我们的兄弟单位,在我们买新的2台溶出仪的同时,也新购买了2台“天津TDTF”的溶出仪,型号都是ZRS-8G的。用了一段时间,也是转杆异响严重,后经厂家调试后正常。另外,是取样装置很奇怪,不是用垫片,而是用一个类似于积木的装置来定取样针的位置,操作人员都说很不习惯。另外,机头跟水浴槽的间距太小,导致取样操作不方便。感觉上没有这家公司以前仪器那么好用。是不是都因为价格竞争太厉害,厂家都不惜降低产品品质来销售。 上面是我在使用溶出仪的一点体会。想抛砖引玉,请大家也谈谈都在哪些品牌的国产溶出仪,说说使用心得和不足之处。据我了解,除了天大天发,国内还有上海黄海、天津新天光等等都有做药物溶出仪,不知道哪位有用过相关产品,也来说说,给我采购一个参考。
我没有顶空进样器,而且现用甲苯做出来的溶剂峰前延严重拖尾,用DB-WAX柱,用80度的柱温。只进化学纯的环氧乙烷是40度而且峰型很好,但是溶解在甲苯里就不出峰了。该怎么办呢
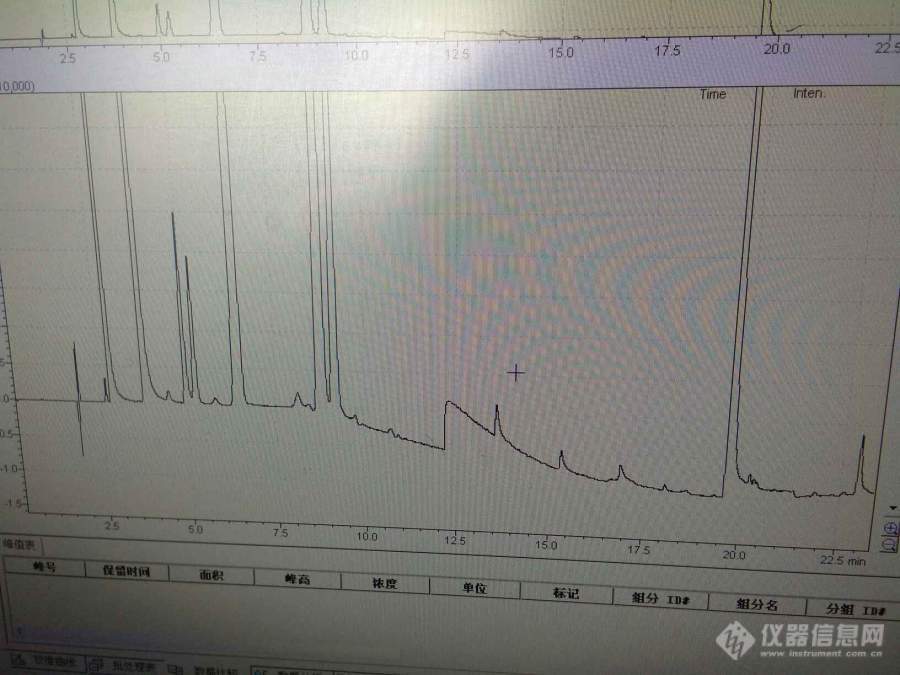
[color=#444444]我想请问一下,[url=https://insevent.instrument.com.cn/t/Mp]气相色谱[/url]中,顶空进样,溶剂为N-甲基吡咯烷酮,柱子DB-624,溶液里面成分有甲醇,乙醇,乙腈,二氯甲烷,正己烷,乙酸乙酯,四氢呋喃,三乙胺,吡啶,DMF,目前三乙胺峰判断不出来,也不确定到底有没有出来峰,图谱已传上,请教一下[/color][color=#444444][img=,690,517]https://ng1.17img.cn/bbsfiles/images/2019/06/201906141009051727_9546_1752329_3.jpg!w690x517.jpg[/img][/color]
最近由于工作需要,在外单位做实验,项目是药物中的溶剂残留,共11种,包括苯。做过类似工作的都知道,苯这玩意儿 最讨厌了,药典中限量是2ppm,因此顶空进样时的溶剂选择很重要,听有经验的师傅说 最好用水做溶剂,灵敏度高,但是还要兼顾其他非极性溶剂,用水就乳化了,郁闷!结合几天的实验,感觉顶空进样 样品处理确实简单,一溶解就行了,并且只要是非挥发性溶剂,哪怕是酸碱 都无所谓,这一点比较好。缺点同样明显,记忆效应明显啊,太容易有残留了,有个残留峰 都 走空白好几针了,依然巨大无比哦,崩溃边缘丶!另外仪器是 目前国内很高端的,具体型号就不说了,气相+顶空+自动进样 70多万啊,不知道是不是最好的,估计 离最贵的不远了!做过类似工作的同行来讨论一下吧!积极参与就可以获得奖励哟
请教大神,用DMSO做溶剂,顶空GCMS检测亚硝胺,不出峰,液体进样没问题,不加溶剂出峰也没问题,麻烦大家帮忙分析分析,谢谢。
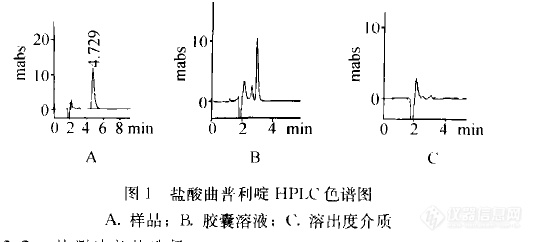
【作者中文名】黎志芳;【作者英文名】Li Zhi-fang (Guangzhou Institute for Drug Control; Guangzhou 510160);【作者单位】广州市药品检验所 广州;【摘要】目的 用HPLC法测定盐酸曲普利啶胶囊中盐酸曲普利啶的溶出量。方法 以Diamonsil C18柱(150 mm×4.6 mm,5μm)为固定相,流动相为甲醇-0.4%醋酸铵溶液(含0.15%三乙胺,用醋酸调pH 7.0)(70:30),流速为1.0 mL·min-1,检测波长为278 nm,进样量40μL,采用外标法定量。结果 盐酸曲普利啶线性范围为1.672-8.363μg·mL-1(r=0.999 9),回收率为100.0%。结论 本方法结果准确,重现性良好,操作简便。用 于测定盐酸曲普利啶胶囊中盐酸曲普利啶的溶出量,较原方法更合理、准确、简便。http://ng1.17img.cn/bbsfiles/images/2012/08/201208061427_381888_2379123_3.jpg
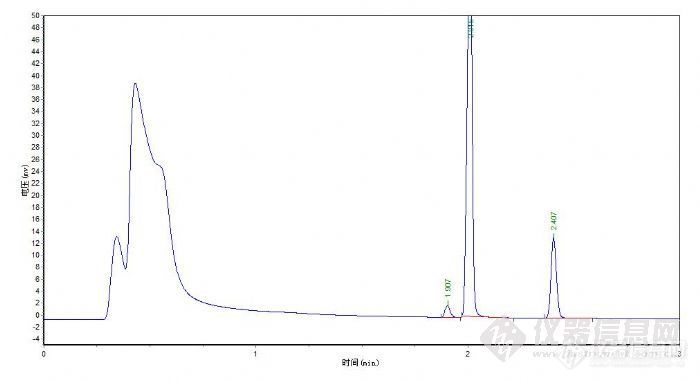
方法:取本品约1.0g,精密称定,置顶空瓶中,精密加水5ml使溶解,密封,作为供试品溶液。精密称取乙腈、丙酮、二氯甲烷适量,用水定量稀释制成每1ml约含乙腈0.41mg、含丙酮5mg、含二氯甲烷0.6mg的混合溶液,精密量取5ml,置顶空瓶中,密封,作为对照品溶液,照残留溶剂测定法(中国药典2005年版二部附录Ⅷ P)测定。以5%苯基-95%二甲基聚硅氧烷(SE-54)(或极性相近)为固定液的毛细管柱为色谱柱,柱温为400C,维持12分钟,进样口温度为2000C,检测器温度为2500C;顶空瓶平衡温度为800C,平衡时间为30分钟。取对照溶液顶空进样,计算数次结果,其相对标准偏差不得过5%。取供试品溶液与对对照品溶液分别顶空进样,记录色谱图,按外标法以峰面积计算,含二氯甲烷不得过0.06%,含乙腈量不得过0.041%。含丙酮不得过0.5%。http://ng1.17img.cn/bbsfiles/images/2011/11/201111011450_327697_2209208_3.jpg检测器用的是FID检测器,出峰顺序为:乙腈,丙酮、二氧甲烷。请各位版友分析一下这是什么原因?1. 在1分钟前出的怪峰,是不是因为顶空瓶压力跟载气上的压力调节得不一致的原因。混合对照时出怪峰,单独进水时不出怪峰?2. 为什么浓度相同的二氯甲烷和乙腈,反倒用顶空进样时,乙脯的峰比二氯甲烷小10倍左右?
我们都知道把固体物质配制成一定浓度的溶液是这样配制的:先在烧杯中称量好样品,计量,然后加入溶剂完全溶解,之后把溶液用玻棒导流至容量瓶,用溶剂洗几次把烧杯里的残液洗进容量瓶中,定容。 但是如果是固体样品易挥发,或者易变质,用烧杯溶解后再转移到容量瓶就不大合适。我觉得可以直接把固体物质放进容量瓶,然后先加入少量溶剂完全溶解,最后定容。技术上是没问题的。就是如果碰到认证的话,这样的操作是否规范? 还有需要抱怨的是:为什么我们所学到的都是必须用烧杯溶解,转移呢,我觉得在转移的过程中更容易出差错,就算非常小心也会有一些些液体流出容量瓶外。特伤脑筋。
求助标准溶液标定,仅需一人平行滴定三次的依据
做残留溶剂时,用安捷伦7820A顶空进样,最近总是峰出来的很小,好像是进样针头处堵了一样,怎么处理?
需要测PLLA微球的溶剂残留,DMF、DMSO、丙酮等常用的溶剂都用过了,现在就发现二氯甲烷和氨水可以溶解至20mg/ml。所以现在就遇到了几个问题[list=1][*]氨水应该不能用,毕竟碱性太强对管路不好;直接进样的话只能用二氯甲烷做空白,但是PLLA微球制备会用到二氯甲烷,所以二氯甲烷也是待测的溶剂之一,终究还是需要用到其他溶剂做空白的[*]样品不溶解的话可以用顶空进样做样吗?现在暂时想试一下用DMF做空白,对照直接用DMF配制(供试品浓度按20mg/ml计算),那我供试品可以直接称100mg加到顶空瓶然后加入5ml的DMF作为供试品溶液吗?[*]因为氨水可以溶样品,所以我想试一下碱性溶液做空白,比如氢氧化钠溶液,氢氧化钠溶液可以作为顶空进样的空白吗?[/list]
如题,请指点一二,不胜感谢!同种溶质使用不同溶剂定容,出峰的时间会受到影响吗?其他的测定条件等都相同。
前提:异丙醇、乙酸乙酯混合标样,甲醇作为溶剂,采用岛津气象色谱顶空进样法,异丙醇不出峰,乙酸乙酯出峰,而同样的混合标样采用直接进样法,二者均可出峰,请问各位大佬,这是为什么呀?为啥异丙醇顶空进样出不来?







 我要推广仪器
我要推广仪器
 下载APP
下载APP