有没有做过HPMC核磁氢谱的啊,我溶在重水中,谱图中只有水峰没有样品峰,烦请有做过HPMC核磁氢谱的指导一下,感谢感谢
吡唑核磁氢谱中,溶剂是DMSO,N上的活泼氢会出峰吗?

核磁氢谱看不懂啊,求高人指点。http://ng1.17img.cn/bbsfiles/images/2012/03/201203301432_358266_2062894_3.jpg结构图核磁氢谱http://ng1.17img.cn/bbsfiles/images/2012/03/201203301433_358267_2062894_3.jpg
有友友测过水凝胶核磁氢谱吗 是测固体核磁还是液体核磁哇 想请教请教
做脉冲核磁氢谱和碳谱一般扫描几次?谢谢!
我们听到核磁共振,可能有人想到的是是医院的核磁、CT之类的,当然医院的核磁和我们所用的核磁原理是差不多的:核磁共振被利用到化学,利用分子结构对氢原子*周围磁场的影响发展了核磁共振谱,后来还有氟谱,13C谱等,核磁共振被利用到医学,水分子的氢原子的核磁共振现象可以被获取到人体内水分子分布的信息,可以精确绘制人体内部结构。 我们今天讲的是应用于化学的核磁共振,氢谱的快速解析。首先我们要知道原理:核磁共振氢谱是根据氢原子的裂分以及化学位移,推断每个基团的氢原子数目和类型,还要知道TMS的作用(相当于内标)。然后我们要知道氚代溶剂的溶剂峰大概出峰位置,比如CDCL[sub]3[/sub]大概在7.26,重水在4.80。“积分”,数氢原子数,然后根据MS提供的分子量或者分子式计算不饱和度。 解析单峰,要知道基团的大概出峰位置,比如-CH3-的出峰大概在1.25的位置,苯环上的氢大概在6-8左右。还要根据一些单峰的特性,比如RCONH2这种,两个氢会分开。比如-CH3-这种可能会形成一个粗的单峰。 我们在解析的时候“不考虑”相互作用,为什么说不考虑呢。因为有很多使氢位置发生变化的效应,比如共轭,比如溶剂效应,比如诱导效应。我们只需要知道很多情况下,该在那个地方的基团出现在其他位置,是合理的就足够了。然后活泼氢,就是与O\S\N相连的氢,如果要证明有活泼氢存在,一定要选择氚代氯仿或者DMSO做溶剂,然后用重水交换。基本上活泼氢的位置不是很固定,不要去考虑,知道有几个活泼氢就可以了。然后根据推导的分子结构与MS等其他仪器进行对照。没学过核磁共振这门课,工作中自学到这些简单的知识,所以是小白解析,仅供参考。
用N-甲基咪唑和一氯甲烷合成的离子液体[Mmin]cl,是种亲水性极强的物质,在做核磁氢谱图时,发现少个氢的峰,经分析可能还是缺少五环上的2位上的氢,请问什么原因,甲基咪唑检测没有问题.
核磁共振技术发展较早,20世纪70年代以前,主要是核磁共振氢谱的研究和应用。70年代以后,随着傅里叶变换波谱仪的诞生,13C—NMR的研究迅速开展。由于1H—NMR的灵敏度高,而且积累的研究资料丰富,因此在结构解析方面1H—NMR的重要性仍强于13C—NMR。解析图谱的步骤1.先观察图谱是否符合要求;①四甲基硅烷的信号是否正常;②杂音大不大;③基线是否平;④积分曲线中没有吸收信号的地方是否平整。如果有问题,解析时要引起注意,最好重新测试图谱。2.区分杂质峰、溶剂峰、旋转边峰(spinning side bands)、13C卫星峰(13C satellite peaks)(1)杂质峰:杂质含量相对样品比例很小,因此杂质峰的峰面积很小,且杂质峰与样品峰之间没有简单整数比的关系,容易区别。(2)溶剂峰:氘代试剂不可能达到100%的同位素纯度(大部分试剂的氘代率为99-99.8%),因此谱图中往往呈现相应的溶剂峰,如CDCL3中的溶剂峰的δ值约为7.27 ppm处。(3)旋转边峰:在测试样品时,样品管在1H-NMR仪中快速旋转,当仪器调节未达到良好工作状态时,会出现旋转边带,即以强谱线为中心,呈现出一对对称的弱峰,称为旋转边峰。(4)13C卫星峰:13C具有磁距,可以与1H偶合产生裂分,称之为13C卫星峰,但由13C的天然丰度只为1.1%,只有氢的强峰才能观察到,一般不会对氢的谱图造成干扰。3.根据积分曲线,观察各信号的相对高度,计算样品化合物分子式中的氢原子数目。可利用可靠的甲基信号或孤立的次甲基信号为标准计算各信号峰的质子数目。4.先解析图中CH3O、CH3N、 、CH3C=O、CH3C=C、CH3-C等孤立的甲基质子信号,然后再解析偶合的甲基质子信号。5.解析羧基、醛基、分子内氢键等低磁场的质子信号。6.解析芳香核上的质子信号。7.比较滴加重水前后测定的图谱,观察有无信号峰消失的现象,了解分子结构中所连活泼氢官能团。8.根据图谱提供信号峰数目、化学位移和偶合常数,解析一级类型图谱。9.解析高级类型图谱峰信号,如黄酮类化合物B环仅4,-位取代时,呈现AA,BB,系统峰信号,二氢黄酮则呈现ABX系统峰信号。10. 如果一维1H-NMR难以解析分子结构,可考虑测试二维核磁共振谱配合解析结构。11. 组合可能的结构式,根据图谱的解析,组合几种可能的结构式。12. 对推出的结构进行指认,即每个官能团上的氢在图谱中都应有相应的归属信号。四. 核磁共振碳谱(13C—NMR)解析图谱的步骤1.鉴别谱图中的非真实信号峰(1)溶剂峰:虽然碳谱不受溶剂中氢的干扰,但为兼顾氢谱的测定及磁场需要,仍常采用氘代试剂作为溶剂,氘代试剂中的碳原子均有相应的峰。(2)杂质峰:杂质含量相对于样品少得多,其峰面积极小,与样品化合物中的碳呈现的峰不成比例。(3)测试条件的影响:测试条件会对所测谱图有较大影响。如脉冲倾斜角较大而脉冲间隔不够长时,往往导致季碳不出峰;扫描宽度不够大时,扫描宽度以外的谱线会折叠到图谱中来;等等,均造成解析图谱的困难。2.不饱和度的计算根据分子式计算的不饱和度,推测图谱烯碳的情况。3.分子对称性的分析若谱线数目等于分子式中碳原子数目,说明分子结构无对称性;若谱线数目小于分子式中碳原子数目,说明分子结构有一定的对称性。此外,化合物中碳原子数目较多时,有些核的化学环境相似,可能δ值产生重叠现象,应予以注意。4.碳原子δ值的分区碳原子大致可分为三个区(1)高δ值区δ>165ppm,属于羰基和叠烯区:①分子结构中,如存在叠峰,除叠烯中有高δ值信号峰外,叠烯两端碳在双键区域还应有信号峰,两种峰同时存在才说明叠烯存在;②δ>200 ppm的信号,只能属于醛、酮类化合物;③160-180ppm的信号峰,则归属于酸、酯、酸酐等类化合物的羰基。(2)中δ值区δ90-160ppm(一般情况δ为100-150ppm)烯、芳环、除叠烯中央碳原子外的其他SP2杂化碳原子、碳氮三键碳原子都在这个区域出峰。(3)低δ值区δ<100ppm,主要脂肪链碳原子区:①不与氧、氮、氟等杂原子相连的饱和的δ值小于55ppm;②炔碳原子δ值在70-100ppm,这是不饱和碳原子的特例。5.碳原子级数的确定由低共振或APT(attached proton test)、DEPT(distortionless enhancement by polarization transfer)等技术可确定碳原子的级数,由此可计算化合物中与碳原子相连的氢原子数。若此数目小于分子式中的氢原子数,二者之差值为化合物中活泼氢的原子数。6.推导可能的结构式先推导出结构单元,并进一步组合成若干可能的结构式。7.对碳谱的指认将碳谱中各信号峰在推出的可能结构式上进行指认,找出各碳谱信号相应的归属,从而在被推导的可能结构式中找出最合理的结构式,即正确的结构式。
各位大虾,谁作过槲皮素的氢谱和碳谱核磁,有谱图的话请上传一下!感激不尽!
求大神指教 海藻酸钠水凝胶核磁氢谱应该测固体核磁还是液体核磁哇 如果液体核磁溶剂应该是什么哇

1、前言笔者刚接触核磁不久,有农药中间体生产企业合成了两种中间体,要鉴定一下合成的物质结构是否正确。两个样品的预期结构如下图所示:[align=center][img=,200,178]http://ng1.17img.cn/bbsfiles/images/2017/07/201707110822_01_3081717_3.png[/img][/align]鉴定物质结构是比较复杂的过程,需要用到多种谱图,综合分析。这里就简单介绍一下核磁共振波谱仪氢谱,使用的仪器为布鲁克600M的核磁共振波谱仪,需把样品(约20mg)溶解到氘代DMSO中,上机测试[img=,400,224]http://ng1.17img.cn/bbsfiles/images/2017/07/201707110823_01_3081717_3.jpg[/img]2、分析步骤样品1所得谱图如下:[align=center][img=,400,374]http://ng1.17img.cn/bbsfiles/images/2017/07/201707110824_01_3081717_3.png[/img][/align][align=left]2.1谱图简析[/align][align=left]2.1.1数峰个数:谱图中,有8个峰,不过根据物质1的结构应该是4组峰(如图,分别标为a/b/c/d),因为,本实验用的拥挤为氘代DMSO,因此化学位移2.5ppm处的应该是DMSO的溶剂峰,同时,经询问客户,样品中有残留溶剂乙醇,需进一步排除乙醇的峰。2.1.2标峰并积分:物质1中含有4组氢,与氧连的-CH[sub]2[/sub][sub],[/sub]由于氧的电负性,使b组氢比a,c组氢的化学位移要大,同时受P的影响,裂分会比较复杂,因此判断4ppm处的多重峰为b组氢,并作为标准,积分为2,然后积分其他峰。4.5ppm处的鼓包,应为残留溶剂乙醇的羟基氢,化学位移3.45ppm四重峰与1ppm三重峰积分面积比为2:3,并且峰的裂分符合乙醇中亚甲基与甲基上的氢的情况,由此,判断3.45ppm与1ppm处是残留溶剂乙醇的峰(杂质峰)。[/align][align=left]2.1.3 排除了溶剂峰与杂质峰后,数谱图上剩余氢的个位数为9,与物质1结构中的氢个数一致。可继续进行分析。[/align][align=left]2.1.4. 对上述9个氢进行归属:6.5-7.5处的两个峰峰面积之和为1,此氢为d氢,由于与P耦合,导致裂分为2个峰;1.49ppm的两重峰为a氢,积分为3,同时受P的耦合也发生了裂分;1.26ppm的三重峰为c氢受临近b氢的耦合分裂成3重峰。同理,得到样品2(不含乙醇)的核磁氢谱,对比两种物质的分子结构,物质2只是比物质1多了两个亚甲基,1.5ppm两侧的积分面积均为2的两个多重峰即符合这两个亚甲基的氢峰。[/align][align=center][img=,400,350]http://ng1.17img.cn/bbsfiles/images/2017/07/201707110836_01_3081717_3.png[/img][/align][align=left]2.2结论简析[/align][align=center]通过以上解析,可见,所测样品1氢谱与物质1吻合,样品2氢谱与物质2吻合。不过,最终确定物质结构还需要13C NMR,31P NMR,及质谱、红外等的佐证。[/align][align=left] 以上是我分析核磁氢谱的一般步骤与思路,因为我没有接受过核磁谱图解析的培训,可能有不妥之处,很多专业术语我也没有完全理解,需要进一步的学习与钻研,写出以上内容也是出于对未知的兴趣,希望给刚刚接触核磁共振谱图的同行一点解谱思路,更希望解谱的高手给予批评与指正。[img]http://simg.instrument.com.cn/bbs/images/default/em09511.gif[/img][/align][align=left][/align]
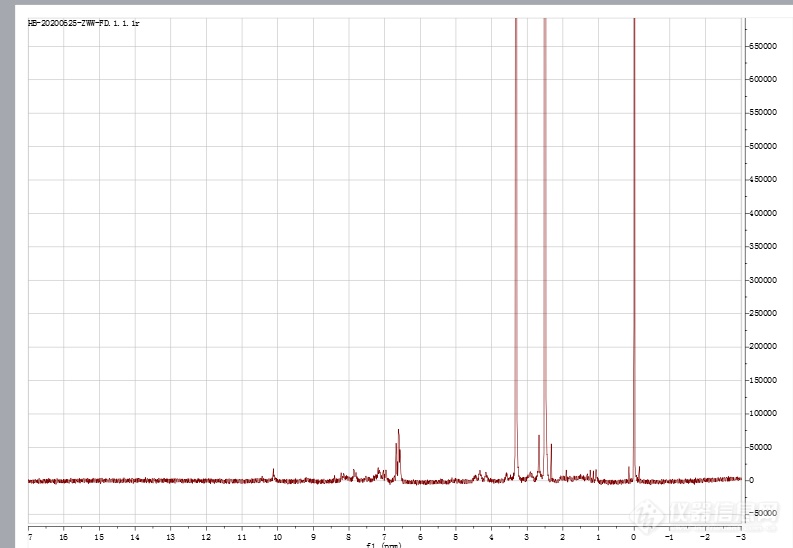
核磁共振氢谱有一个文件显示的信号强度很大,有好几万而且左上角显示的文件名称后缀为.1r。平时做的核磁信号强度只有几百而且左上角文件名后缀是.fid。不知道是怎么回事,这两个有什么不一样吗?求解答。[img=,690,476]https://ng1.17img.cn/bbsfiles/images/2020/06/202006281219419928_3647_3535854_3.png!w690x476.jpg[/img][img=,690,498]https://ng1.17img.cn/bbsfiles/images/2020/06/202006281220238022_6220_3535854_3.png!w690x498.jpg[/img]
高校实验室欲购核磁共振波谱仪,400M,能作固体样品,请专家推荐几款,先谢谢了
核磁共振检测请教高人,我要做PBT和PBT的接枝物的核磁共振氢谱。请问浓度多大为好啊?
如题 偶尔会遇到核磁氢谱上面出现一个倒峰 一般是因为什么,谢谢!
[em09501] 各位老师好,我需要做一个800兆的核磁共振氢谱,请问去哪里做?一般花多少钱?谢谢各位!
有谁知道核磁谱图中,与硼连接的氢原子的化学位移啊,望大神告知!实验做到这一步卡了,若大神知道,还望告知,定不胜感激!
核磁碳谱的灵敏度要低于氢谱,原因是13C的自然丰度只有1.1%,13C的磁旋比(γ)只有1H的1/4,信号强度与磁旋比立方成比例。 脉冲傅立叶变换核磁共振波谱仪特点 (1)不采用固定频率扫描或固定磁场扫频的方法,而是将某一范围内的不同频率同时提供给样品。 (2)样品至于固定磁场中,并接受短脉冲电磁辐射的照射,脉冲辐射的频率分布范围,正适合置于给定磁场中的碳核共振所需的频率
在使用核磁共振氢谱检测较复杂的化合物时,如果用脂肪氢的积分值为标准,通常会出现芳香区的积分偏低的情况.而当结构简单时,则不明显.请问这是什么原因造成的?有没有办法校正?谢谢.
《核磁共振氢谱分析》-电子书[~94838~]
求聚丙烯酸丁酯的核磁氢谱,谢谢!
如题:怎样通过核磁共振氢谱算dr值?
新手求助分析一张氢谱核磁共振图,真心不懂啊。所用的溶解试剂是CDCL3,可能混合有少量的甲醇,根据之前的实验分析,该物质应该较大,分子量在600左右,含有烯烃。帮忙分析,定当感谢!
求:软脂酸的核磁共振氢谱图
若核磁管无法spin,氢谱能否拿到满意的结果
RT 可以相信chemdraw模拟的核磁氢谱吗
在做有机化学相关的课题时,最常用的仪器之一就是核磁共振波谱仪,通过该仪器能够获得相关有机化合物的信息。其中核磁共振氢谱是最常见的手段。要做出一张比较完美的谱图不仅需要按照标准要求制备样品,也需要后期利用核磁解析软件对原始数据进行解析,在这个过程中有时会遇到很多问题,需要根据不同的情况进行处理。首先,第一步是利用软件打开原始数据,本人习惯用MesReNova软件,一下就以该软件为例分享一些小小的经验。一般选择fid文件,打开数据后,观察信号峰是否大小合适,如果不合适,可选择Fit to Height选项,使得信号峰处于合适的高度,如果调整之后依然不理想,可以通过将鼠标放在信号峰区域,滚动鼠标来调整信号峰处于最佳的高度。然后,观察一下谱图的基线是否平整,如果不平整,需要选择Baseline Correction进行调整,一般选择自动调整,若自动调整的效果不佳则选择手动调整,直到基线平整为止。同时,观察各个信号峰是否有上下飘动的情况,如果有,需要选择Phase Correction进行调整,如有必要选择手动调整,进入手动调整后,需要先调整PH0的值,调整好之后再调整PH1的值,知道信号峰的峰型达到正常的状态。需要注意的是,一般以氘水作溶剂的时候,这种情况很常见,需要耐心仔细的进行调整,如果实在达不到最佳状态,有时可以通过增大化合物的浓度重新做核磁来改善这种情况。接下来,需要选择Reference选项,标注一下所用氘代试剂的标准化学位移值,具体的标准数值参照这篇文献:[i]J. Org. Chem. [/i][b]1997, [/b][i]62, [/i]7512-7515。在接下来,选择Peak Picking选项对信号峰的化学位移进行标注,需要注意的是,有些时候氘代试剂会含有少量的水或者化合物中混有溶剂,需要通过对照不同的氘代试剂中这些干扰物的信号峰的位置从而进行仔细甄别。另外,软件默认的字体一般比较小,需要点击鼠标右键,选择Properties, 在Peaks选项栏中选择字体及其合适的大小。最后,选择Integration对相关信号峰进行积分,计算每个信号峰对应的质子的数量,从而推断出化合物的结构。有时,需要计算耦合常数,则选择Multiplets Analysis进行计算,可以自动计算,不过手动计算的结果更准确,然后导出耦合常数的信息。同样的,需要点击鼠标右键,选择Properties, 在Integrals选项栏中选择字体及其合适的大小。这就是从原始数据到一张完整的核磁共振氢谱图的解析过程,都是本人的一些经验,希望对大家有帮助。
请教高人,我要做PBT和PBT的接枝物的核磁氢谱。请问浓度多大为好啊?
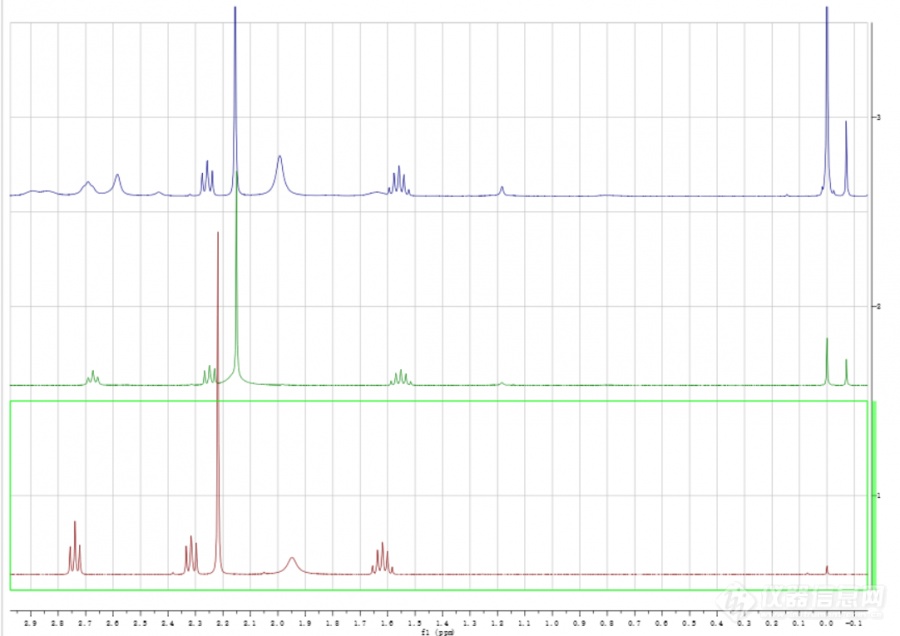
[color=#444444]请求帮助!做的核磁氢谱,溶剂为氘代氯仿,里面含有一定百分比的四甲基硅烷,每个样加入的溶剂体积和样品体积一样多,可是为什么7.26的溶剂峰高和0处的四甲基硅烷峰高差别很大,分别拉到一样大后放到一起还是这样,并且出现了位移为负值的峰。请问水峰会出吗?叔胺N和伯胺N上的H分别会出峰吗?请求大神帮忙分析[/color][color=#444444][img=,690,487]https://ng1.17img.cn/bbsfiles/images/2019/05/201905161413219196_9493_1701336_3.png!w690x487.jpg[/img][img=,690,501]https://ng1.17img.cn/bbsfiles/images/2019/05/201905161413230617_626_1701336_3.png!w690x501.jpg[/img][/color]
做核磁氢谱用时,常用的内标是TMS,我看有文献在做四硝基甘脲时用HDMS作内标,请问它们各自的使用条件是什么?有什么区别?







 我要推广仪器
我要推广仪器
 下载APP
下载APP