促肾上腺皮质激素ACTH(18-39)抗体现货促销
【详细说明】:促肾上腺皮质激素ACTH(18-39)抗体【浓 度】:1mg/1ml 抗体来源【宿 主】:兔源、鼠源、其他 克隆:单克隆抗体、多克隆抗体【适 用】:Human, Mouse, Rat, Chicken, Dog, Pig, Cow, Horse, Sheep, Monkey, others。 抗体类型:一抗 研究领域:细胞生物、神经生物学等 【性 状】:促肾上腺皮质激素ACTH(18-39)抗体冻干粉或液体【相关标记】:FITC、Gold 、HRP、PE PE-Cy3、PE-CY5、PE-CY5.5 、PE-CY7 、RBITC 、 Alexa Fluor 350、Alexa Fluor 488 、 Alexa Fluor 555 、Alexa Fluor 647、AP 、APC 、Biotin 、Cy3 、Cy5 、Cy5.5 、Cy7 。【储 存 液】: Preservative: 15mM Sodium Azide, Constituents: 1% BSA, 0.01M PBS, pH 7.4 or PBS with 0.1% sodium azide and 50% glycerol pH 7.3. -20oC, Avoid freeze / thaw cycles.【产品应用】 :Immunohistochemistry (IHC), Flow Cytometry (FACS) , Western Blotting (WB) , ELISA , Immunohistochemistry , Immunohistochemistry (Paraffin-embedded Sections) (IHC (p)) , Immunoprecipitation (IP) , Immunocytochemistry (ICC) ,Immunofluorescence (IF)等。促肾上腺皮质激素ACTH(18-39)抗体ADCY8 腺苷酸环化酶8抗体 (1)IgG :血清中含量最高,因此是最重要的抗感染分子,包括抗菌、抗病毒、抗毒素等。 IgG 还能激活补体,结合并增强巨噬细胞的吞噬功能(调理作用和 ADCC 效应),穿过胎盘,保护胎儿及新生婴儿免受感染。 (2)IgA :分单体和双体两种。前者存在血清中,后者存在于黏膜表面及分泌液中,是黏膜局部抗感染的重要因素。(3)IgM :是分子量最大,体内受感染后最早产生的抗体,具有很强的激活补体和调理作用,因此是重要的抗感染因子,且常用于诊断早期感染。 (4)IgD :主要存在于成熟 B 细胞表面,是 B 细胞识别抗原的受体。 (5)IgE :血清中含量最少的抗体,某些过敏性体质的人血清中可检测到,参与介导 I 型超敏反应和抗寄生虫感染。促肾上腺皮质激素ACTH(18-39)抗体现货促销中,为您推荐相关优质检测抗体:Anti-Leptin receptor(long) 瘦素受体抗体(长) Anti-Leptin receptor(long) 瘦素受体抗体(长) Anti-Lgr5/GPR49 肠上皮干细胞蛋白抗体 Anti-LH (Mouse Anti-Human Luteinizing Hormone Monoclonal Antibody) 鼠抗人促黄体生成素抗体 Anti-L-HDC (L-Histidine decarboxylase) L-组氨酸脱羧酶抗体 hu, mo, rat, bov, dog, pig, chi Anti-LHRH/GNRH (luteinizing hormone-releasing hormone) 黄体激素释放激素抗体/促性腺激素释放激素抗体 Anti-LIF (leukemia inhibitory factor) 白血病抑制因子抗体 Anti-Lingo-1 Nogo受体作用蛋白抗体 Anti-Livin (Inhibitors of apoptosis proterins Livin) 一种新的凋亡抑制蛋白抗体 anti-LFABP/FABP-1(Liver Fatty acid binding protein) 肝脏型脂肪酸结合蛋白抗体 anti-LFABP/FABP-1(Liver Fatty acid binding protein) 肝脏型脂肪酸结合蛋白抗体 Anti-LN (laminin) 层粘连蛋白抗体 Anti-Lpin1 protein Lpin1 抗体 Anti-Lpin1 protein Lpin1 抗体 Anti-LRP/MVP (Lung resistance related protein) 肺耐药相关蛋白抗体 Anti-LRRK2 (Leucine-rich repeat kinase 2) 帕金森氏病致病基因/神经系统新功能基因抗体 Anti-Lumbrokinase 抗蚯蚓纤溶酶抗体/抗蚓激酶抗体 Anti-Lysozyme 溶菌酶抗体 anti-LYVE-1(lymphalic vessel endotheilial hyaluronan receptor 1) 淋巴管内皮透明质酸受体抗体 Anti-M2-PK ( pyruvate Kinase M2) 丙酮酸激酶-M2抗体 Anti-M2-PK (pyruvate Kinase M2) 丙酮酸激酶-M2(小鼠来源抗体) Anti-Integrin αM/CD11b (Mac-1/CR3A)(Integrin-alpha2) 巨噬细胞表面分子/整合素-α2抗体 Anti-ChRM1 (muscarinic acetylcholine receptor) 毒蕈碱型乙酰胆碱受体M1抗体 Anti-MADCAM-1(-Mucosal addressin cellular adhesion molecule-1) 粘膜选址素抗体 Anti-MAG-a/b (Myelin associated glycoprotein L / S -MAG ) 髓鞘相关糖蛋白a/b抗体 Anti-MAG-a/L-MAG (Myelin associated glycoprotein) 髓鞘相关糖蛋白-a抗体 Anti-MAGE-1/HLA-A1 protein (melanoma antigen family A member 1) 黑素瘤抗原-1抗体 Anti-MAPKK1 (MAP kinase kinase 1) 丝裂原活化蛋白激酶激酶1 Anti-MAPKK2 (MAP kinase kinase 2) 丝裂原活化蛋白激酶激酶2抗体 Anti-Maspin (mammary serine protease inhibitor) 抑癌基因抗体 Anti-Matriptase 蛋白裂解酶(一种新的癌基因)抗体 Anti-MBP (Myelin Basic Protein, MBP) 髓鞘碱性蛋白抗体 Anti-MCP-1 (monocyte chemotactic protein1) 巨噬细胞趋化蛋白-1抗体 Anti-M-CSF (Macrophage Colony Stimulating Factors) 巨噬细胞克隆刺激因子抗体 Anti-MDM2 (urine double minute 2) 双微体2癌基因抗体 Anti-Megsin/SER—PINB7 丝氨酸(或半胱氨酸)蛋白酶抑制剂B7抗体 Anti-Melan-A/MART-1 黑色素瘤相关抗原/黑色素-A抗体 Anti-Metal ion transporter 拟南介金属离子转运蛋白抗体 Anti-Mfn1 (Mitofusin1) 线粒体融合蛋白1抗体 Anti-MGMT (O6-methylguanine-DNA methyltransferase) O6甲基鸟嘌呤DNA甲基转移酶抗体 anti-MT(metallothionein) 金属基质硫蛋白抗体 anti-MGr1-Ag/37LRP(P37-kDa laminin receptor precursor)(NT) 层粘连蛋白受体1抗体(N端) anti-MGr1-Ag/37LRP(P37-kDa laminin receptor precursor)(CT) 层粘连蛋白受体1抗体(C端) Anti-MICA(MHC class I polypeptide-related sequence A) 一种细胞应激分子抗体 Anti-Midnolin isoform Protein 1 中脑核仁蛋白1抗体 Anti-Midnolin isoform Protein 2 中脑核仁蛋白2抗体 Anti-MIF (Macrophage Migration Inhibitory Factor) 巨噬细胞移动抑制因子抗体 Anti-MIP-1α (macrophage inflammatory protein 1α) 巨噬细胞炎症因子1α抗体 Anti-MIP-1β (macrophage inflammatory protein 1β) 巨噬细胞炎症因子1β 抗体 Anti-MMP-1(matrix metalloproteinases-1) 基质金属蛋白酶-1抗体 Anti-MMP-1(matrix metalloproteinases-1)anti-Mouse 基质金属蛋白酶-1抗体(小鼠) Anti-MMP-13 (Matrix metalloproteinase 13) 基质金属蛋白酶13抗体 Anti-MMP-14(Matrix metalloproteinase-14) 基质金属蛋白酶-14抗体 Anti-MMP-2(Collagenase IV /Gelatinase A/Metallo proteinase-2) 基质金属蛋白酶-2抗体 Anti-MMP-3(matrix metalloproteinase-3/Transin-1/SL-1/Stromelysin-1 precursor) 基质金属蛋白酶-3抗体 Anti-MMP-7(Matrilysin/matrix metalloproteinases-7) 基质金属蛋白酶-7抗体 Anti-MMP-9(matrix metalloproteinase 9) 基质金属蛋白酶-9抗体 Anti-β-2-MG 鼠抗人β2微球蛋白抗体(单抗) Anti-Mo anti-KLH 小鼠抗血蓝蛋白抗体 Anti-MOG (myelin oligo-dendrocyte glycoprotein-MOG) 髓鞘少树突胶质细胞糖蛋白抗体 Anti-Mouse anti-human HAS 鼠抗人血清白蛋白单克隆抗体 Anti-Mouse IgA 兔抗小鼠IgA抗体 Anti-MPO (myeloperoxidase) 髓过氧化物酶抗体 Anti-MRP1(Multidrug Resistanec-Associated Protein 1) 多药耐药相关蛋白1抗体 Anti-MRP2 (multidrug resistance-associated protein2) 多药耐药相关蛋白2抗体 Anti-MRP3(Multidrug Resistanec-Associated Protein 3) 多药耐药相关蛋白3抗体 Anti-MrpL28 (mitochondrial ribosomal protein L28) 线粒体核糖体蛋白L28抗体 Anti-MSH-2 (MutS homolog 2) 错配修复蛋白2抗体 anti-MLH1(Mutl homolog l gene) 错配修复蛋白1抗体 Anti-MSLN (mesothelin) 间皮素抗体 anti-MUC5AC/Mucin 5AC(Gastric Mucin M1) 胃粘液素抗体 Anti-MTR-1A (Melatonin receptor-1A) 褪黑素受体/松果体素受体抗体 Anti-mucin-1/Muc-1/CD227 antigen (Epithelial Membrane Antigen ) 粘蛋白-1/上皮膜抗原抗体 Anti-MyD88 (myeloid differential protein-88) 髓样分化蛋白抗体 Anti-Myelin P0 protein( peripheral myelin prothein Zero MPZ MPP) 外周髓磷脂P0蛋白/P0蛋白抗体 Anti-Myosin (Smooth Muscle) 鼠抗人心肌肌凝蛋白(平滑肌) 单抗 Anti-N-AChR α4 (Nicotinic-Acetylcholine receptor α4) 烟碱型乙酰胆碱受体α4抗体 Anti-N-AChR α7 (Nicotinic-Acetylcholine receptor α7) 烟碱型乙酰胆碱受体α7抗体 Anti-Nanog 胚胎干细胞关键蛋白抗体 anti-Natrexone 抗纳曲酮抗体IgG Anti-NAP1 (nucleosome assembly protein 1) 核小体组装蛋白1抗体 Anti-N-cadherin N-钙粘附分子抗体 Anti-N-coR1 (Nuclear receptor co-repressor 1) 核受体辅助抑制因子抗体 Anti-Nephrin Protein 肾病蛋白抗体 Anti-Nestin 巢蛋白/神经上皮干细胞蛋白抗体 Anti-Nestin 巢蛋白/神经上皮干细胞蛋白抗体 Anti-Neurobeachin protein (AKAP550) 蛋白激酶锚定蛋白/激酶固定蛋白抗体 Anti-Neurocan 神经粘蛋白抗体 Anti-Neurofascin-155 神经束蛋白-155 Anti-NF-H(Neurofilament triplet H) 高分子量神经丝蛋白抗体 Anti-NFKBp65(p65 NF-kappa B p65NFKB) 细胞核因子/k基因结合核因子抗体 Anti-NF-L(Neurofilament triplet L) 低分子量神经丝蛋白抗体 Anti-NF-M (Neurofilament triplet M) 中分子量神经丝蛋白抗体 Anti-NF-κBp50(p50 NF-kappa B p50NFKB) 细胞核因子50/κ基因结合核因子50抗体 Anti-NGF-R/p75NTR/CD271(p75 Neurotrophin R) 神经生长因子受体抗体 Anti-NGF-β 神经生长因子-β抗体 anti-NGN3(neurogenin 3 Neurog3) 神经元素3抗体 Anti-NGX6 (nasopharyngeal carcinoma/NPC associated gene 6) 鼻咽癌细胞相关基因6抗体 Anti-NHE1(Na+/H+ Exchanger) 钠氢通道蛋白抗体 Anti-NIK(NF-kappaB-Inducing Kinase) NFkB诱导的激酶抗体 Anti-NIS(Na+/I-symporter) 钠碘转运体蛋白抗体 Anti-NK-1/SuRCtance P Receptor (Neurokinin receptor1 Tachykinin receptor1) P物质受体抗体

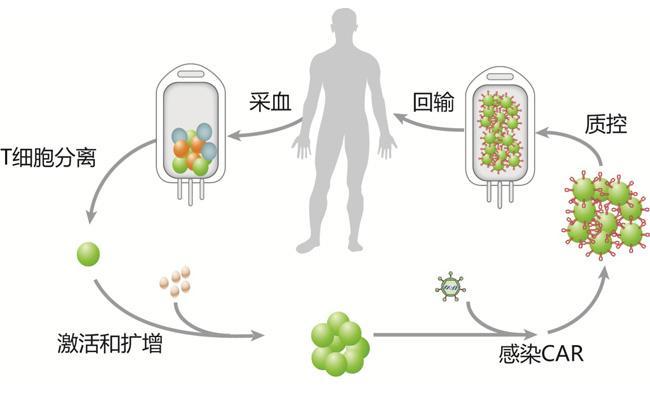


























 我要推广仪器
我要推广仪器
 下载APP
下载APP