推荐厂家
暂无
暂无
 留言咨询
留言咨询
 400-860-5168转4839
留言咨询
留言咨询
400-860-5168转4839
留言咨询
留言咨询
 400-873-7896
留言咨询
400-873-7896
留言咨询
 400-860-8560
留言咨询
400-860-8560
留言咨询
 400-860-5168转4438
留言咨询
400-860-5168转4438
留言咨询






1971年Kleppe等人在Journal of molecular biology上发表文章首次准确、精炼、客观的阐述了PCR方法,1976年一种从嗜热水生菌(Thermus aquaticus)分离得到的热稳定的DNA依赖的DNA聚合酶的应用大大增加了PCR的效率。而现今所发展出来的PCR则是源于由Saiki和Mullis等人于1988年发表在Science上的一篇论文,Mullis当时服务于Perkin Elmer(PE)公司,因此PE公司在PCR界有着特殊的地位。后来PE被Applied Biosystems Inc.(ABI)公司收购、分拆、再转卖,而PCR的专利和倍受信赖的PCR仪器生产和销售就留在ABI名下。到如今,PCR方法愈发趋向自动化,并从中衍生出更多的新技术方法,可以说,PCR技术是支撑现代分子生物学发展的一块重要基石。这种技术的广泛应用催生了一个庞大的市场,多个公司均有各种类型的商品化PCR仪出售。PCR的专利目前依然掌握在ABI和Roche(罗氏)两大公司手中,去年业界颇为引人瞩目ABI诉MJ公司侵犯侵犯PCR仪知识产权案最终以MJ败诉并宣布破产、最终被Bio-rad收购暂告一段落。其后还会不会有后继的故事还需拭目以待。 PCR原理 DNA的半保留复制是生物进化和传代的重要途径。双链DNA在多种酶的作用下可以变性解链成单链,在DNA聚合酶的作用下,以单链为模版,根据碱基互补配对原则复制成新的单链,与模版配对成为双链分子挎贝。在体外实验中发现,DNA在高温时也可以发生变性解链,当温度降低后又可以复性成为双链。因此,通过温度变化控制DNA的变性和复性,并设计与模板DNA的5’端结合的两条引物,加入DNA聚合酶、dNTP就可以完成特定基因的体外复制,多次重复“变性解链—退火—合成延伸”的循环就可以以几何级数大量扩增特定的基因。 发现耐热DNA聚合酶对于PCR的应用有里程碑的意义,该类酶可以耐受90℃以上的高温而不失活,不需要每个循环加酶,使PCR技术变得非常简捷、同时也大大降低了成本,PCR技术得以大量应用,并逐步应用于临床。 从PCR原理可以看出,PCR仪的关键是升降温的步骤。现在偶尔还能听到一些前辈们笑谈早年的PCR实验如何在3个水浴锅中完成的趣闻。经过不断改进,今天的PCR已经越来越完善和智能化。出于市场推广的战略需要,各厂家的PCR仪型号不同,着力宣传的技术指标和参数也不尽统一,编者在这里简单列出选购时我们认为应该考虑的常用指标,希望有助于大家选购PCR仪的选购技巧。PCR仪介绍及其选购 PCR仪的种类总体来说可以分为两大类:PCR扩增仪和实时荧光定量PCR仪,普通的PCR扩增仪又衍生出带梯度PCR功能的梯度PCR仪、和带原位扩增功能的原位PCR仪等等。1996年由ABI公司首先推出将扩增和检测融为一体的实时荧光定量PCR仪,此后很多公司如Eppendorf、ROCHE、Corbett、MJ等等都先后推出不同款式的定量PCR仪。
问题1:不出现扩增条带 引物:引物质量是PCR失败或扩增条带不理想、容易弥散的常见原因。对策为:①选定合适的引物合成单位;②引物的浓度不仅要看OD值,最好用引物原液做琼脂糖凝胶电泳,如一条引物有条带,一条引物无条带,此时做PCR有可能失败,应与引物合成单位协商解决,如一条引物亮度高,一条引物亮度低,在稀释引物时要平衡其浓度;③引物应高浓度小量分装保存,防止多次冻融或长期放冰箱冷藏而导致变质降解失效;④引物设计不合理,如引物长度不够,引物之间形成二聚体等。 Mg2+浓度:Mg2+离子浓度对PCR扩增效率影响很大,浓度过高可降低扩增的特异性,浓度过低则影响PCR扩增产量甚至使PCR扩增失败而不出扩增条带。 靶序列变异:如靶序列发生突变或缺失,影响引物与模板特异性结合,或因靶序列某段缺失使引物与模板失去互补序列,其PCR扩增是不会成功的。问题2:出现非特异性扩增带 PCR扩增后出现的条带与预计的大小不一致,或大或小,或者同时出现特异性扩增带与非特异性扩增带。非特异性条带出现的原因:一是引物与靶序列不完全互补,或引物聚合形成二聚体;二是Mg2+离子浓度过高、退火温度过低,及PCR循环次数过多有关。 其次是酶的质和量,往往一些来源的酶易出现非特异条带而另一来源的酶则不出现,酶量过多有时也会出现非特异性扩增。对策为:必要时重新设计引物;减低酶量或调换另一来源的酶;降低引物量,适当增加模板量,减少循环次数;适当提高退火温度或采用二温度点法(93℃变性,65℃左右退火与延伸)。问题3:出现片状拖带或涂抹带 PCR扩增有时出现涂抹带或片状带或地毯样带。可能由于Mg2+浓度过高,退火温度过低,循环次数过多,GC含量过高引起。对策为:适当降低Mg2+浓度;增加模板量;减少循环次数;加入添加剂甘油或Qiagen公司的Q-solution。问题4:污染的监测——对照试验 1.阳性对照:应设有阳性对照,它是PCR反应是否成功、产物条带位置及大小是否合乎理论要求的一个重要标志。阳性对照要选择扩增度中等、重复性好,经各种鉴定是该产物的标本,如以重组质粒为阳性对照,其含量宜低不宜高(100个拷贝以下)。 2.阴性对照:每次PCR实验务必做阴性对照。它包括①标本对照:被检的标本是血清就用鉴定后的正常血清作对照,被检的标本是组织细胞就用相应的组织细胞作对照;②试剂对照:在PCR试剂中不加模板DNA或RNA进行PCR扩增,以监测试剂是否污染。 3.重复性试验 4.选择不同区域的引物进行PCR扩增问题5:污染的处理 (一)环境污染 1.稀酸处理法 2.紫外照射(UV)法 (二)反应液污染,可采用下列方法之一处理: 1.DNase I法 2.内切酶法 3.紫外照射法 4.Gama射线辐射法 (三)尿嘧啶糖苷酶(UNG)法 由于UV照射的去污染作用对500bp以下的片段效果不好,而临床用于检测的PCR扩增片段通常为300bp左右,因此UNG的预防作用日益受到重视和肯定。 (四)固相捕获法 用于去除标本中污染的核酸和杂质。 (五)RS-PCR法(RNA-specificPCR) 也称为链特异性PCR,主要指用于RNA模板的特异性PCR法,该法可明显降低假阳性而不影响PCR的敏感性。 (六)抗污染引物法 该对引物扩增时通过病毒DNA克隆如入质粒的位点,这一区域只存在于完整的原病毒中。如果重组质粒污染了标本,就不能扩增出任何条带,即使出现了扩增带,其大小也与预期的不同。只有原病毒DNA才能被引物扩增,因此只要出现预期大小的扩增带就可以证明标本是阳性的,该法试用于环状靶分子系列。
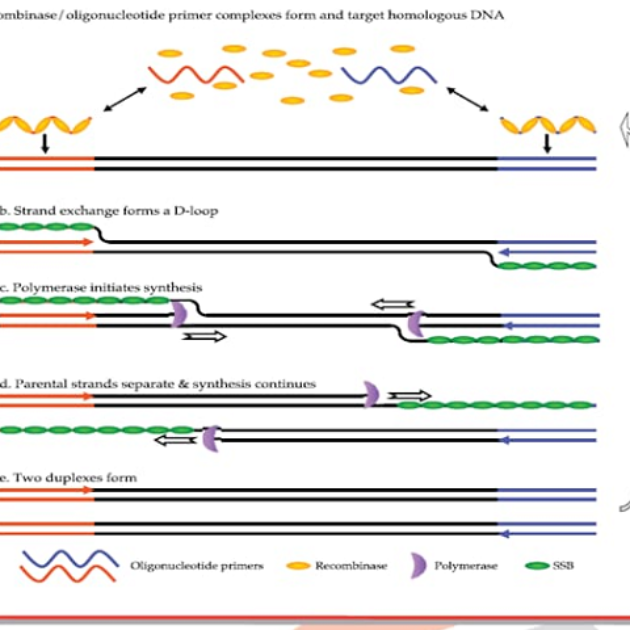
什么是PCR技术? PCR(Polymerase Chain Reaction)技术,中文译为聚合酶链式反应,是一种在体外(试管、切片…)扩增核酸的技术。PCR的基本反应包括以DNA为模板的反应和以mRNA为模板的反应。 PCR技术是模拟体内天然DNA(脱氧核糖核酸)的复制过程。以扩增DNA为例,其基本原理是在模板、引物、4种dNTP和耐热DNA聚合酶存在的条件下,特异扩增位于两段已知序列之间的DNA区段的酶促合成反应。







 我要推广仪器
我要推广仪器
 下载APP
下载APP