

视频号

抖音号

哔哩哔哩号

前沿资讯手机看

中山大学药学院教授,博士生导师。主要从事生物质谱新技术的开发及应用。



分享到微信朋友圈
打开微信,点击底部的“发现”,
使用“扫一扫”即可将网页分享到朋友圈。
在生物学的中心法则中,信息从DNA流向RNA,最终转化为执行生物学功能的蛋白质。由于遗传变异、RNA可变剪接和翻译后修饰(PTMs)的存在,蛋白质形成了多样的蛋白分子形式(proteoforms)。目前,Top-Down 蛋白质组学(TDP)已经成为了全面研究蛋白分子形式的最强大技术,它通过Top-Down质谱(TDMS)的实验,不需要酶切,直接分析完整的蛋白质,以提供蛋白分子形式的整体视图。TDMS既需要对完整蛋白的精确质量分析(Top),也需要控制下游气相离子的碎裂来获取序列的信息和PTMs的位点(Down)。与TDP不同,Bottom-Up蛋白质组学(BUP)通过分析酶切后的肽段(通常小于3 KDa)来获取蛋白质的信息,由于多肽相对于完整蛋白更加容易分离、电离和断裂,BUP有着比TDP更加广泛的应用。但是由于BUP获取的肽段数量有限,因此提供的序列覆盖普遍偏低,这就导致了蛋白分子形式信息的丢失。此外,相较于TDP来说,BUP无法提供组合的PTMs信息。(如图1)
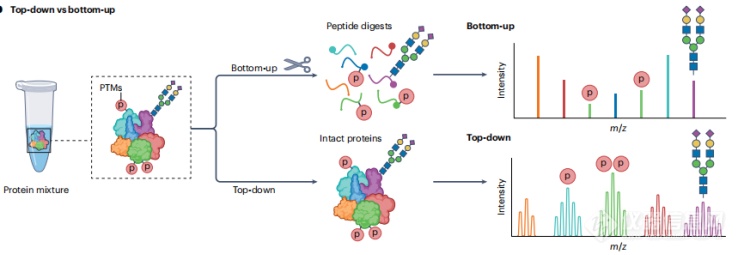
图1. TDP和BUP的对比
TDP的基本实验流程如图2所示,包括前端样品制备和分离,完整质量分析和碎片分析,以及关于蛋白分子形式的鉴定表征和定量的信息学。
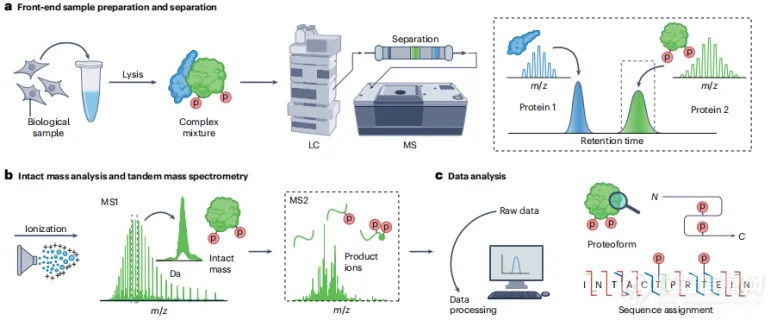
图2. TDP的基本工作流程
样品制备
传统的蛋白质提取方法使用Good’s缓冲液,它具有高盐浓度(>100 mM),蛋白酶抑制剂、磷酸酶抑制剂和表面活性剂,用于总蛋白质的溶解。这些常规试剂往往与TDP不相容,因为它们会干扰蛋白质离子检测并抑制质谱信号。不相容的盐和小分子可以通过超滤管离心或使用尺寸排除色谱(SEC)自旋柱去除。在样品制备过程中应注意尽量减少人工引入的蛋白分子形式。例如,蛋白酶和磷酸酶抑制剂通常包括在萃取缓冲液中,以尽量减少体外蛋白质降解和去磷酸化。蛋白应该低温保存,以减慢任何的修饰反应。表面活性剂能促进疏水膜蛋白的细胞渗透和增溶,但是表面活性剂会对蛋白的质谱分析造成信号抑制。蛋白质沉淀法通常使用氯仿/甲醇混合物或丙酮,可以去除表面活性剂和其他质谱不相容的污染物。然而,蛋白质沉淀方法耗时且可能导致蛋白质损失、实验变异性或溶解性问题。因此,可切割表面活性剂已被开发出来,如可酸降解的Rapigest、ProteaseMAX、MaSDeS;可光降解的4-己基苯基偶氮磺酸酯;可氧化还原降解的N-十二烷基二硫-β-d-麦芽糖苷等。
样品分离和富集
前端分离和富集策略可以选择性地分离亚蛋白质组,在质谱分析之前从复杂的生物样品中捕获和富集低丰度蛋白质。例如,可以通过差速离心的方式获取细胞器,蛋白再从细胞器中提取。另一种方式是通过亲和纯化的方式,基于抗体的方式已经在完整蛋白的靶向分析上得到了应用,但是该方法受到高特异性和高质量抗体需求的限制。为了解决这些挑战,可特异性捕获蛋白的表面功能化的多价超顺磁性纳米颗粒和集成纳米蛋白质组学的方法被开发出来。
仪器
早期的TDP实验依靠单四极杆和三四极杆(分别为Q和QqQ)质谱仪进行完整蛋白分析。这些系统具有较差的质量分辨能力,使得电荷状态测定困难,并且有限的质量电荷比(m/z)范围导致对大蛋白质的适用性较低。
高质量分辨能力对于TDP尤为重要,因为完整蛋白质产生的片段离子可以产生卷曲的质谱,其中具有不同电荷态的各种离子可以部分重叠。许多现代质谱仪器可以可靠地实现高分辨率,包括傅里叶变换质谱系统,如离子回旋共振(FTICR)和Orbitrap质谱仪,以及飞行时间(TOF)和四极杆-飞行时间 (Q-TOF)仪器。
完整蛋白的分离
蛋白质组的复杂性给TDP带来了巨大的挑战,需要在质谱分析之前分离完整的蛋白质。当处理较大的蛋白质(≥30 kDa)时,这一挑战尤其明显,因为随着蛋白质大小的增加,ESI质谱中的离子信号迅速减少。早期的分离方法使用凝胶电泳技术,例如二维凝胶电泳分离、虚拟的二维凝胶电泳分离、直接利用MALDI MS的干胶法和PEPPI-MS等。还可以通过SEC(尺寸排阻色谱)、RPLC(反相液相色谱)、HIC(疏水相互作用色谱)、IEX(离子交换色谱)以及多维的LC方法来分离。例如一个3D LC方法,通过耦合HIC - IEX - RPC,相较于2D IEX -RPLC MS将蛋白质的鉴定数目提高了14倍。此外,CE-MS的最新进展可以用于变性和非变性的TDP分离。离子淌度质谱(IMS)是基于分子在电场作用下的气相运输性质和碰撞截面积(CCS)分离蛋白质,高分辨率的IMS已被证明可以用于分离高序列同源性的蛋白。
串联质谱技术
串联质谱(MS/MS)技术,在TDP中通常包括通过选择前体蛋白离子,将其解离成更小的片段离子并分析片段离子,从而得出蛋白质的初级结构和修饰(如图3a)。有多种活化/解离方法可用于生成产物离子。大多数仪器可以进行碰撞诱导离解(CID),也称为碰撞激活离解,通过与中性气体分子(如氮气或氩气)相互作用的碰撞激活来产生b/y离子(图3b)。红外多光子解离(IRMPD)涉及吸收低能红外光子产生b/y离子,并可能在吸收多光子后产生二级和高阶片段离子,从而产生更广泛的蛋白质序列信息。基于电子的解离方法(ExD),如电子捕获解离(ECD)和电子转移离解(ETD),在产生高序列覆盖率方面往往优于CID。ExD产生的c/z产物可用于确定的蛋白质形态表征和PTM定位。使用193 nm或213 nm激光,紫外光解离(UVPD)可以生成更复杂的串联质谱,序列覆盖率与ExD方法相当或更高。此外,结合四极质量过滤器、线性离子阱和Orbitrap的混合平台可以进行质子转移电荷还原(PTCR),以简化产物离子谱。
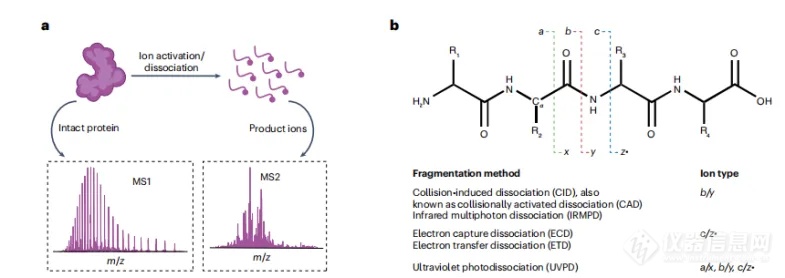
图3. a, 串联质谱技术示意图。b, 不同解离方式产生的碎片离子。
数据采集
可采集的谱图数量取决于仪器的占空比和峰的宽度。最常见的TDP数据采集方法是数据依赖的采集(DDA)。在DDA中,收集一个完整的质谱扫描,并选择几个前体离子(通常是最丰富的)进行片段化。与数据非依赖的采集(DIA),即不分离前体离子的质谱扫描碎片,正在迅速发展并在BUP工作流程中采用,为TDP提供了令人兴奋的机会。
结果处理
TDP的数据信息丰富但是解析难度大,考虑到同位素和电荷态的影响,以及人类蛋白质组的高动态范围,使得谱图分析和对于低丰度蛋白的检测更加困难。TDP的谱图往往有更加复杂的同位素包络,通常不会观察到单同位素峰。大多数工具依赖于Averagine模型来解同位素并预测理论同位素分布。当谱图不能进行同位素分解时,谱图去卷积可以使用多个电荷态离子来推导出一种蛋白分子形式的平均质量。
目前正在开发用于存储质谱数据的标准化文件格式。最通用的文件格式是mzML(最新版本1.1.1),这是一种由人类蛋白质组组织蛋白质组学标准倡议(HUPO-PSI)支持的XML格式。几个开源软件库可以转换,读取和写入质谱文件格式,包括ProteoWizard,JmzML,mzJava和pymzML。
获得去卷积谱图后的下一步是针对蛋白质或蛋白质序列数据库搜索去卷积质谱,以识别具有错误发现率(FDR)控制的蛋白分子形式并表征PTMs,具体的搜索原理的概述可以查看原文Results-data analysis部分,在这里不再赘述。最后,定量分析不同样品间丰富度的差异。数据库可以使用来自UniProt、RefSeq、GENCODE或相关资源的蛋白质序列数据库。这些数据库只包含序列,不包含PTMs的信息。可以根据PTMs位点和类型,构建可用的数据库,但要注意限制PTMs的可变数目,否则产生的组合数据会过于庞大。DNA或RNA-seq数据可用于构建具有样品特异性基因突变和备选剪接事件的蛋白分子形式序列数据库。
定性与定量分析
TDP提供了对蛋白分子形式的全面了解,使鉴定、新蛋白分子形式的发现和深入的序列表征成为可能。TDP具有独特的优势,因为它可以表征组合PTMs与多基因家族中不同基因编码的同型异构体,这些异构体通常具有高序列同源性。例如,肉瘤蛋白具有多种亚型和PTMs,如N端二甲基化、乙酰化、磷酸化和甲基化。单个肌肉细胞的蛋白质形态变化可以通过TDP进行研究。当单个蛋白分子上存在多个PTMs时,TDP是唯一可以解析复杂蛋白形态和组合PTMs的技术。例如,TDP可以鉴定组蛋白的多种蛋白分子形式,以及定量描述PTMs之间的化学计量学。
TDP的定量方式和BUP类似,主要有非标记定量(label free):采用不同蛋白分子形式的信号强度定量;同位素标记(isotope labeling):采用不同分子量的同位素标记来定量;化学标记(chemical labeling):采用化学报告基团来定量,尤其是在MS2水平上。(如图4)其他标记技术——如氨基酸稳定同位素标记(SILAC)、同量异位(isoabric)标记、假同量异位(pseudoisobaric)标记和NeuCode SILAC——已经显示出定量TDP的潜力。
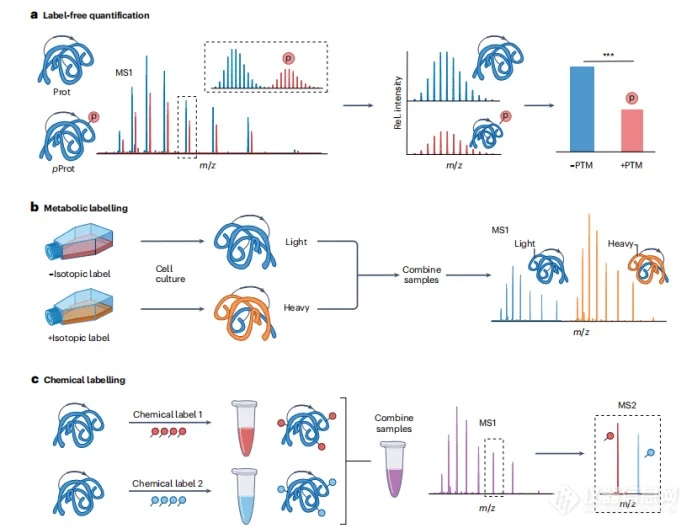
图4. 不同定量方法
统计学分析和错误率计算
TDP的软件通常会使用E值和P值来反映串联质谱和蛋白分子形式的匹配程度,此外FDR值也常被用来描述鉴定的可靠性。对于定量的TDP.对于定量TDP分析,统计分析通常使用单向方差分析和Stundent’s 检验(双尾)。多次测试调整通常使用benjamin - hochberg方法进行。如有必要,可采用非参数Kruskal-Wallis单因素方差分析和Wilcoxon秩和检验进行组间比较。对于人类临床样本的定量TDP,随机截距的线性混合效应模型可以进一步表征人类个体之间的异质性。作者还介绍了一些TDP软件,例如,TopPIC、MSPathFinder、TopMG和pTop等。
应用
作者在这一部分列举了许多相关研究,考虑到篇幅限制,感兴趣的读者可以在原文中的Application部分查看。主要包括(1)全球蛋白分子形式的发现。(2)癌症:例如,一项全球TDP研究从结直肠癌细胞的2332种蛋白质中鉴定出23000多种蛋白分子形式,并揭示了转移性和非转移性细胞之间蛋白分子形式水平的巨大差异。(3)心血管疾病:将蛋白质组学应用于心脏生物学和临床诊断已经取得了进展。例如,TDP分析了“CARDIA研究”中的配对血清样本,揭示了载脂蛋白AI和AII与心脏代谢指标之间的蛋白分子形式特异性关联。(4)神经退行性疾病:失调的PTMs可以影响神经退行性疾病中的蛋白质聚集,许多PTMs是神经退行性疾病中蛋白质病变的调节剂。例如,阿尔茨海默病受到β或tau淀粉样蛋白磷酸化和β淀粉样蛋白中异天冬氨酸形成的影响。(5)传染病。(6)生物制剂:例如单克隆抗体,抗体偶联药物等基于蛋白的药物。(7)临床TDP,例如,使用MALDI-TOF-MS鉴定病原体,它可以直接从完整的细菌细胞表面快速检测到蛋白分子形式。
重现性与数据存储
TDP是一个相对较新的领域,与成熟的BUP方法不同,普遍接受的实验方法和数据报告标准尚未制定。由CTDP(Consortium for TDP)领导的标准化工作正在推动实验室间的比较,以更好地了解挑战并提高重现性。蛋白分子形式易受样品处理和仪器方法变化的影响,因此科学的严谨性和充分的数据报告实践非常重要。
所有TDP数据都应公开提供。许多期刊已经实现了这一要求,但这需要共同的努力来确保正确的数据处理和报告实践得到执行。CTDP的Proteoform Repository为科学家提供了一个独特的中心,可以浏览存储的蛋白分子形式并提供TDP数据集。数据存储库对于TDP数据遵守FAIR数据存储标准是必不可少的。将TDP数据集平台化并作为中央存储库的新途径和举措将对促进TDP数据的可访问性和共享非常有价值,这反过来将使TDP领域受益。
面临的挑战与优化策略
TDP的新技术在不断地发展,但是仍旧面临着诸多的挑战:(1)灵敏度。传统的TDP工作流程需要大量的样品(微克级的总蛋白或者数百万的细胞),以获得高质量的数据。新的高灵敏度的方法正在开发,例如CE-MS可实现对于单个细胞的检测,Nanopots技术可用于TDP,还有可以提高蛋白提取率的表面活性剂与尿素联用。(2)高分子量的蛋白质分子形式。高分子量的蛋白质难分离、信号差、仪器负担大。这就需要超高分辨率的平台,如FTICR质谱仪。在质谱分析之前,基于SEC或凝胶技术的基于尺寸的分离方法,例如,整合蛋白质组学方法或PEPPI-MS,可以解决大离子分析的挑战。但是没有单一的分离策略或者MS/MS仪器可以分析整个蛋白质组,需要开发新方法、新仪器以及改进的信息学工具来克服。(3)蛋白质的串联质谱。蛋白质在序列末端的片段化效率较高,而在中间的片段化覆盖率有限。这种差异在较大的蛋白质中更为明显,并且被认为是由于在变性条件下仍然存在的。内部碎片离子是由至少两个骨干断裂产生的,没有N或C端,在TDP碎片离子中越来越多地被考虑。最近开发的TDP软件ClipsMS可以将内部片段质量分配给蛋白质序列,从而提高整体覆盖深度。(4)定位修饰位点。由于不稳定的PTMs,低丰度的蛋白形式,PTMs的实验定位和蛋白质分子形式化学成分的精确表征具有挑战性。富集策略可以增强低化学计量或低丰度信号;还需要优化特定的碎片方法,例如,通过使用更温和的基于电子的方法,如ETD或ECD。(5)通量问题。TDP相对较低的吞吐量和较高的数据复杂性是新老用户的主要障碍。基于发现的TDP数据处理包括去卷积和数据库搜索,这可能需要几个小时到几天,具体取决于软件性能和搜索参数。自动制备和分离系统的发展,以及软件性能的提升都有助于改善通量的问题。
展望
TDP是目前唯一能够确定蛋白质形分子形式特征并量化其丰度的技术。由于蛋白质分子形式的基本重要性及其作为细胞、环境或生物系统健康标志的潜在作用,TDP技术有望继续快速发展。需要解决的两个关键领域是改进复杂蛋白质分子形式混合物的深度表征和大分子量蛋白的识别和表征。一个令人兴奋的发展是单离子测量,它可以在现有的商业仪器和专门的前体类型上实现。液相色谱固定相、CE-MS、基于IMS的分离的发展以及与多维方法的整合将继续改善蛋白质组学的测量。利用基因组、转录组以及BUP的信息,可以构建更精准的数据库,用于分析一些更复杂的PTMs,例如蛋白的糖基化,这也是TDP的一个优化方向。为了将蛋白质分子形式与相关的可测量输出(例如转录物和代谢物)联系起来,并破译生物学的基本原理,可以将多组学的测量结合起来。此外,通过结合微流体、质谱成像和单离子测量等先进技术,有望将单细胞和空间生物学扩展到蛋白质分子形式分析。
本文发表在Nature Reviews Methods Primers 上,Top-Down Proteomics[1] ,通讯作者是美国威斯康辛大学麦迪逊分校化学系的Lloyd M. Smith和Ying Ge教授。文章介绍了Top-Down蛋白质组学的基本工作流程、应用以及面对的挑战。
[1] Roberts, D.S., Loo, J.A., Tsybin, Y.O., et al. Top-down proteomics[J]. Nature Reviews Methods Primers,2024,4(1):38.
[来源:仪器信息网] 未经授权不得转载
 对于NGS,谁是下一个NIPT?
对于NGS,谁是下一个NIPT?
蛋白质组学大咖Robert Moritz:π-HuB计划的推出恰逢其时
2024.06.20

2024.11.01

2024.11.01

275项仪器类服务项目入选!上海市公布第三批仪器类科技创新券拟兑付名单
2024.11.01

2024.11.01

2024.11.01
品牌合作伙伴






























版权与免责声明:
① 凡本网注明"来源:仪器信息网"的所有作品,版权均属于仪器信息网,未经本网授权不得转载、摘编或利用其它方式使用。已获本网授权的作品,应在授权范围内使用,并注明"来源:仪器信息网"。违者本网将追究相关法律责任。
② 本网凡注明"来源:xxx(非本网)"的作品,均转载自其它媒体,转载目的在于传递更多信息,并不代表本网赞同其观点和对其真实性负责,且不承担此类作品侵权行为的直接责任及连带责任。如其他媒体、网站或个人从本网下载使用,必须保留本网注明的"稿件来源",并自负版权等法律责任。
③ 如涉及作品内容、版权等问题,请在作品发表之日起两周内与本网联系,否则视为默认仪器信息网有权转载。
![]() 谢谢您的赞赏,您的鼓励是我前进的动力~
谢谢您的赞赏,您的鼓励是我前进的动力~
打赏失败了~
评论成功+4积分
评论成功,积分获取达到限制
![]() 投票成功~
投票成功~
投票失败了~









