
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转1639
仪器信息网认证电话,请放心拨打
【复旦大学Engineering案例】采用ACR连续多级搅拌反应器、CSTR、固定床、连续膜分离装置等连续流合成瑞德西韦的核碱基单元
连续流合成瑞德西韦的核碱基单元
【背景介绍】
2019年严重急性呼吸综合征冠状病毒(SARS-CoV-2)引起的冠状肺炎(COVID-19)造成了全球毁灭性的公共卫生危机,瑞德西韦(Remdesivir)引起了世界的关注。这种磷酸核苷酸类似物被称为Veklury [1,2],由于专家对其有效性存在不同的观点[1,2,4],目前其仍在全球范围内进行单独或与其他药物联合使用的一系列临床试验,以期治疗感染冠状病毒的患者。在第一波新冠病毒大流行期间,瑞德西韦在美国和日本获准作为紧急药物使用,同时,在欧盟、新加坡、澳大利亚、韩国和加拿大获准有条件使用权限[1],允许住院的成人和儿童冠状疾病患者使用瑞德西韦治疗。
迄今为止,冠状病毒已在全世界造成约1.75亿人感染,380万人死亡[5]。更糟糕的是,全球确诊的病例数量仍在上升,一些更具传染性的变异株在世界各地引发了新的警报[6,7]。鉴于全球规模的病毒流行,以及生产瑞德西韦所需的活性药物成分(API)的巨大潜在需求,确保这种化合物的充足供应可能是一个关键问题[8]。尽管科研工作者已经在改进瑞德西韦的合成方法方面付出了很大的努力,但目前使用的间歇式批量生产仍然是一个漫长的、资源密集型的过程,必须按顺序完成,并且存在难以大规模操作、产量低的缺点[9]。
复杂的间歇过程似乎会影响在冠状病毒大流行等紧急情况下快速生产大量瑞德西韦的能力。此外,间歇式生产会导致高昂的成本,因此限制了这种药物的广泛使用[9]。因此,需要开发一种高效且可扩展的合成方案来制备瑞德西韦。通过逆合成分析(图1),瑞德西韦可以由三种不同的结构单元组成,包括核糖内酯单元(2)、具有立体生成磷中心的磷酰胺单元(3)和类似腺嘌呤的碱基单元(4;即7-卤代吡咯[2,1-f][1,2,4]三嗪-4-胺)[10,11]。
由于在后期进行有效地执行关键的C-糖基化步骤需要大量的核碱基单元(4),复旦大学手性分子工程中心的陈芬儿院士课题组在连续流系统中高效且经济地合成了核碱基单元(4),旨在开发一种独特的安全、高效、可放大的连续流系统,从而实现快速、可放大地制备瑞德西韦。该方案优化了涉及危险和不稳定中间体的反应,很好地控制放热反应,并且显著增强了液-液两相反应。此外,低温反应也得到了很好的调控。后续处理过程完全集合到了反应序列中,形成了一个整体的全连续流动系统,最大限度地提高了整体工艺效率。

图1. 瑞德西韦的逆合成分析
前期研究已经报道了几种合成路线来获得关键中间体吡咯[2,1-f][1,2,4]三嗪-4-胺(5)[12–16]。通过化合物5的简单溴化或碘化即可得到目标化合物4。在此基础上,本文描述了7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(X=Br时为4)的合成步骤。O'Connor等人[12]和Dixon等人[13]揭示了一种通过2,5-二甲氧基四氢呋喃与叔丁基氨基甲酸酯反应来提供NH-t-丁氧基羰基(BOC)保护的1-氨基吡咯的路线。随后与氯磺酰异氰酸酯和N,N-二甲基甲酰胺(DMF)进行氰化反应,得到NH-BOC保护的1-氨基-1H-吡咯-2-碳腈。BOC脱保护,然后在回流的乙醇中与乙酸甲脒(FAA)和磷酸钾环化,得到所需的中间体5 [12,13]。
Knapp等人[14]报道了一个类似的方案,其中5-二甲氧基四氢呋喃通过与2-氨基异吲哚-1,3-二酮或碳氮叔丁酯反应转化为1-氨基吡咯或NH-BOC保护的1-氨基吡咯。在甲酸中用NaOAc处理1-氨基吡咯,或在甲酸中用醋酐处理NH-BOC保护的1-氨基吡咯,均可产生N-(1H-吡咯-1-基)甲酰胺(N-(1H-pyrrol-1-yl)formamide)。随后与氰胺缩合得到N′-氰基-N-(1H-吡咯-1-基)甲酰胺;在刘易斯酸介导的环化后,得到化合物5 [14]。Patil等人[15]报道了一种两步法,其中吡咯-2-甲醛在水中用羟胺-O-磺酸(HOSA)和KOH处理,生成1-氨基-1H-吡咯-2-碳腈。
然后在碳酸钾存在下,在回流乙醇中用FAA处理色谱纯化后的1-氨基-1H 吡咯-2-碳腈,得到5[15]。然而,这些方法的实际应用受到起始原料的有限可用性和相对较高成本,以及分离相关中间体所需的快速柱层析的要求等限制。Paymode等人[16]最近报道了一种方法,首先通过使用磷酰氯(POCl3)和DMF的Vilsmeier–Haack反应对吡咯进行甲酰化。随后,通过与羟胺、醋酐和吡啶反应,将生成的吡咯-2-甲醛氧化转化为吡咯-2-碳腈。随后的N-氨基化和环合反应得到所需的5。这种方法的优点是使用了低廉的起始材料。
然而,POCl3的使用会导致含磷废水的产生,可能对环境产生严重的有害影响。同时,使用羟胺、醋酸酐和吡啶(3.5–5.0当量)进行氧化转化不仅增加了产品纯化的难度,还增加了制备5的总成本。此外,氯胺直接N-氨基化反应需要复杂的氯胺萃取和浓缩过程才可以提高产量,这将导致萃取溶剂和能源的大规模消耗。基于之前的研究,本文改进了合成方案,以无色谱纯化的方式从廉价且容易获得的吡咯中获得核碱基中间体4,如图2所示。首先通过Vilsmeier-Haack反应将吡咯(6)转化为吡咯-2-甲醛(7)。
本文没有使用传统的Vilsmeier–Haack试剂POCl3/DMF,而是使用双(三氯甲基)碳酸盐(BTC)和DMF来形成Vilsmeier盐。因此避免了含磷废水的产生。随后,在水中用较低成本的HOSA处理吡咯-2-甲醛(7),可以完成甲酰基官能团的氧化转化,从而获得吡咯-2-碳腈(8)。然后与O-(二苯基膦基)羟胺(DPPH)进行N-胺化,并与FAA环化,得到关键中间体吡咯[2,1-f][1,2,4]三嗪-4-胺(5)。最后,用N-溴代琥珀酰亚胺(NBS)溴化得到目标化合物7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4),该方案具有较高的区域选择性和产率。

图2. 改进后合成化合物4路线
从实用的角度来看,传统的间歇釜式方法在生产放大时可能会受到限制。首先,Vilsmeier–Haack和N-氨基化反应是高度放热的反应,并且都涉及危险化学品,在批量条件下很难控制。通常这种类型的反应都是在分批模式下进行的,即在低温下缓慢地向反应混合物中添加一种试剂,以防止由于釜式反应器中混合不足和散热缓慢而出现失控情况[17]。该操作适用于小规模的间歇过程,但由于比表面积降低可能需要过长的物料添加时间,不适合大规模生产。因此,大规模的釜式生产过程具有耗时、低效的缺点。
此外,由于快速反应和混合不足,如何保证釜式反应器中的温度均匀是一个重大挑战[18,19]。因此,可能会引发副反应和进一步的二次反应,导致产量损失和安全问题。其次,吡咯-2-甲醛(7)氧化转化为吡咯-2-碳三腈(8)是在不互溶的液-液两相混合物中进行的。传质速率在两相反应中起着重要的作用,在两相反应中,整体的反应速率受两相间活性物质转移速率的影响。因此,在传统间歇反应器中,氧化反应受到低效率的质量传递的影响,只有在有限的表面体积比(高达约2000 m2∙M−3)[20,21]中才可以实现,导致表观动力学受限。
此外,间歇式反应器中的两相流体动力学是基于反应器形状、尺寸、叶轮结构、搅拌速度等的一个众所周知的复杂函数。这使得反应器的放大变得十分困难,通常直到反应动力学和液-液流体动力学得到充分理解后才可以进行大规模的生产。第三,为确保形成目标化合物4的高选择性,溴化反应需要严格的低温条件(从−20到−78°C)。对于传统的间歇式反应器来说,有效且一致地维持低温是很困难的,因为当将间歇式反应器扩大到更大尺寸时,体积的增加速度远远快于外表面面积[22]。因此,间歇式反应器的低比表面积从根本上限制了生产的规模。
最后,间歇釜式过程由于是分步操作,涉及多步的反应序列、分离和纯化步骤[23]。通常,在每个合成反应完成后,需要将产物从反应混合物中进行分离并纯化,然后将获得的纯产物用于下一个反应。这种方法是劳动密集型、资源密集型、耗时且浪费资源[23]。最终,从起始原料到最终API产品(如瑞德西韦)可能需要长达12个月的时间,并且在不同的操作阶段需要大量的中间体库存[9]。
近年来,制药行业出现了向连续制造发展的趋势[25,26]。微反应器的使用在推动间歇式工艺向连续流工艺过渡方面发挥了重要的作用[27,28]。与传统的间歇式反应器相比,微反应器提供了独特的优势:小反应体积、快速混合和良好的质量和热传递,从而提高了反应性能和安全性,和对反应变量进行更精确的控制[29]。此外,与间歇过程相比,连续过程的生产放大要容易很多,可以通过对流动装置进行重复并联或放大反应体积来增加生产通量[29]。此外,通过使用连续流技术,可以将多个合成步骤集合成一个流线型生产体系,并且可以避免分离和中间体纯化的过程[30]。因此,使用最先进的连续流技术可以节省大量资源、空间、时间和能源。
本文计划通过采用连续流动技术克服间歇釜式合成的上述限制。本文目标是开发一种独特的安全、高效、可放大的连续流系统进行目标化合物4的合成。该方案优化了涉及危险和不稳定中间体的反应,很好地控制放热反应,并且显著增强了液-液两相反应。此外,低温反应也得到了很好的调控。后续处理过程完全集合到了反应序列中,形成了一个整体的全连续流动系统,最大限度地提高了整体工艺效率。
【实验方法】
反应器I、反应器II、反应器IV和反应器V均是由内径(ID)为0.6 mm、外径(OD)为1.6 mm的全氟烷氧基(PFA)管构成。反应器III是一个连续搅拌反应设备(ACR;AM Technology,UK),其特点是一个安装在横向振动电机上的哈氏合金反应模块[31]。反应模块的中心流板包含反应室、互连通道和搅拌器。反应室由一系列约9.8 mL的反应单元组成,每个单元内有一个可自由移动的搅拌器。单个反应单元由长度为30 mm、宽度为4 mm的互连通道连接。当反应模块被振动电机振动时,搅拌器在单元中进行快速反转的横向运动[32]。
从原理上看,反应室模拟了毫升级连续搅拌釜式反应器(CSTR)的级联,以实现塞流特性。有关ACR的更多详细信息,可以参考其用户手册[31]。另外,本工作中使用最多的的是T型微型混合器来实现流体的混合。研究发现,对于混相流体,使用简单的T型微型混合器(步骤1、4和5)可以实现良好的混合。然而,当两股流体不相溶时,使用T型微混合器会导致反应流的不完全混合。
因此,在第2步中,采用了一种所谓的两相错流微混合器(CFMM)来混合不相容液体。此外,本文中还使用了其他几种类型的设备和装置用于将处理程序整合到反应序列中,包括气液分离器(GLS)、连续流固体过滤器(CFSF)、微型连续搅拌釜式反应器(m-CSTR)、液-液膜分离器(LLMS)、环形离心萃取器(ACE)和固定床反应器(FBR)。本文中使用的连续流设备和装置的详细说明见附录A.S3。
本文中使用注射器泵(Fusion 101、Fusion 200、Fusion 4000、Fusion 6000;Chemyx,美国)或高效液相色谱(HPLC)泵(SF1005A,Sanotac,中国)用于泵送溶液。反应料浆使用蠕动泵(德国Masterflex 77200-60型)泵送。背压调节器(BPR)购自Chemtrix(荷兰)。文中使用标准的1/4〃-28单向阀(美国IDEX Health&Science)防止逆流。流体连接采用标准的1/4〃-28螺纹接头,配有1/16〃盘管和卡套(中国Runzefluidsystem)。
【结果与讨论】
第一步涉及吡咯(6)与DMF和BTC的Vilsmeier–Haack反应,从而生成吡咯-2-甲醛(7)。DMF和BTC之间的接触导致快速形成Vilsmeier复合物,该复合物具有热不稳定性,加热时可快速产生高温和高压[33,34]。这在大规模操作时容易导致安全问题[18,19]。因此,在间歇模式下,这种转化可以通过在0°C下缓慢地将BTC溶液加入吡咯和DMF的混合物中来实现[17]。添加完成后,将反应混合物加热至更高温度(45°C)进行进一步的反应。在间歇模式下完成毫摩尔级别的反应需要5个多小时(详细说明见附录A. S2.1)。
作者最初尝试在反应器I出口没有安装GLS的情况下进行流动反应,结果发现,6到7的转化率为95%,两股反应物(1.0当量吡咯,1.05当量DMF,0.35当量BTC,DCE)通过T型微混合器混合(图3中的步骤1),理论停留时间为30分钟(即反应器I的内部体积除以总流速)。然而,作者观察到流动反应器中会快速形成气体,使反应混合物的流速不稳定,导致实际停留时间无法控制。
为了解决这个问题,作者在反应器I之后连接了一个GLS,氮气(N2)通过加压气体入口进入GLS,并在其气体出口安装了一个可调节的背压阀(BPR)。在这种方法中,反应器I在BPR控制的设定压力下由N2稳定加压。然后将从GLS排出的反应混合物与进入的饱和Na2CO3水溶液一起流进m-CSTR-1(图3),可以促进中间产物盐的水解,从而获得甲酰化合物7。

图3. 两步连续流动合成吡咯-2-碳腈(8)的示意图。P1–P8为泵。PYL:吡咯;V:反应器的内部容积;T:反应器温度;tR:停留时间;rt:室温。
基于改进后的连续流系统,本文通过进一步的优化实验(表2)发现,35°C的较低温度和3 bar(1 bar=105 Pa)的背压可以实现在5分钟的停留时间内完成6到7的完全转化。
表2. 在反应器I中连续流动合成7的优化实验

FP1:P1泵的流量;FP2:P2泵的流量;P: 背压。
a当停留时间不同时,摩尔比保持不变。
b通过GC/MS峰面积百分比确定6的转化率。
完成生产化合物7的优化连续流方案之后,本文接下来计划将流程中的步骤2集合到步骤1中(图3)。随着反应器m-CSTR-1中形成固体盐,收集到的料浆被继续泵入到分离器CFSF-1中,用以去除可能堵塞下游反应器II的固体。在步骤2中,吡咯-2-碳醛(7)与HOSA发生氧化反应生成吡咯-2-碳腈(8)。在初步实验中,来自CFSF-1和HOSA水溶液的滤液通过两相CFMM直接流入反应器II。然而该方案并不令人满意,最佳结果化合物7只有79%的转化率,停留时间为30分钟。
CFSF-1滤液中水相的存在可能会影响后续步骤2中7的反应活性。因此,作者在CFSF-1之后加入了分离设备LLMS-1。滤液通过m-CSTR-2输送至LLMS-1,以连续的方式去除水相。从LLMS-1流出的有机相通过CFMM与HOSA水溶液混合,所得混合物进入反应器II。经过条件优化后,该优化方案在室温下以5分钟的停留时间将化合物7完全转化为8(表3)。
表3. 在反应器II中连续流动合成8的优化实验

FP5: P5泵的流量; FP6: P6泵的流量
a当停留时间不同时,摩尔比保持不变。
b 通过GC/MS峰面积百分比确定7的转化率。
接下来,反应器II的流出料与饱和Na2CO3水溶液一起流入到反应器m-CSTR-3中。由于固体盐是在m-CSTR-3中形成的,因此该料浆混合物随后通过CFSF-2以去除会堵塞LLMS-2的固体。LLMS-2可以快速分离两相混合物中的有机相。该优化后的两步反应-分离一体化连续流过程得到的化合物8分离收率为47.6%,处理量为3.02g·h−1。
第3步反应涉及了吡咯-2-碳腈(8)的N-氨基化,从而获得1-氨基-1H-吡咯-2-碳腈(9)。
该反应中有NaH的参与,尤其是在大规模生产条件下,会带来潜在的安全隐患。因此,釜式反应是通过将干燥THF中的底物8缓慢添加到NaH–THF混合物中,然后在THF中逐滴添加Ph2P(O)ONH2来实现的(详细说明见附录A. S2.3)。因此,釜式生产操作的效率和生产率都很低。由于NaH和Ph2P(O)ONH2不溶于四氢呋喃,并且产生了固体副产物,这些对将该反应转化为基于微通道的流动过程造成了阻碍。为了解决这个问题,本文使用了Coflore ACR反应器来促进料浆的流动。
考虑到要将ACR直接集合到之前两步流动序列的复杂性,作者首先考察了进行纯化吡咯-2-碳腈(8)的单步连续流动反应(图4)。将两股反应物(THF中1.3当量NaH、THF中1.2当量Ph2P(O)ONH2和1.0当量的化合物8)以特定的流速泵入ACR连续多级搅拌反应器中。作者发现化学计量比和停留时间对该连续流反应结果具有很大的影响,相关实验如表4所示。

图4. 单步连续流合成化合物9,P9–P14为泵

英国AM Technology 连续多级搅拌反应器ACR图片
表4. 在反应器III中连续流动合成9的优化实验

FP9:P9泵的流量;FP10:P10泵的流量。
a因为形成的气体影响,这是测量后的实际停留时间。
b 8的转化率由GC峰面积百分比确定。
结果表明,当NaH与Ph2P(O)ONH2的摩尔比小于1.17时,尽管停留时间相对较长,为16分钟,但化合物8最大转化率为50%。此外,当采用较高的摩尔比为1.63(NaH:Ph2P(O)ONH2)时,等量的化合物8仅在6分钟内就实现了完全转化。在最优连续流条件下,温度30℃、停留时间6min,最终获得了化合物9的分离产率为97%。值得注意的是,该反应在连续流中的反应时间仅为6分钟,而在间歇过程中则需要超过5小时(详细对比见附录A. S2.3)。此外,ACR连续多级搅拌反应器能够很容易地保持连续10小时以上的运行。因此,证明了使用ACR续多级搅拌反应器可以成功地进行混合料浆的连续流动过程。
随后,本文研究了从6合成9的三步全连续流过程。由于反应器II的流出物是DCE和水的液-液两相混合物,水的存在无疑会使NaH失活,从而阻碍步骤3的反应。因此,CFSF-2和LLMS-2依次被集合到反应器II之后(图5),以期对反应器II的输出物料进行连续过滤分离处理;然后,滤液流入LLMS-2分离出有机相。按照第3步的连续流优化方案,LLMS-2的有机相与THF中的Ph2P(O)ONH2(1.2当量)和THF中的NaH(1.3当量)一起被泵入ACR中,即可以实现9的三步全连续流动合成。
然后,通过CFSF-3进行连续过滤ACR流出的悬浮液。滤液与饱和NH4Cl水溶液一起流入m-CSTR-5中。然后通过ACE-1和EtOAc泵送出混合物,最终将粗产物9连续萃取获得有机相。ACE-1的有机相随后被泵入RE-1,经过减压浓缩快速去除溶剂。三步连续流合成化合物9的分离产率为44.3%,总停留时间为34min,产量为2.6g·h-1。

图5. 三步全连续流合成1-氨基-1H-吡咯-2-碳三腈(9)的示意图
在第4步反应中,1-氨基-1H-吡咯-2-碳三腈(9)在碱的催化下与FAA发生环化反应,得到吡咯[2,1-f][1,2,4]三嗪-4-胺(5)。前期的釜式环化反应是在85°C条件下,在乙醇中用纯化后的9与FAA和K2CO3反应进行的。该反应需要10小时才能达到完全转化(详细说明见附录A. S2.4)。由于K2CO3在乙醇中是以固体形式存在,作者尝试将K2CO3装填到FBR中,并泵送FAA溶液通过FBR以便将间歇过程转化为连续流。
然而,结果表明在这种模式下不能发生反应。然后作者利用1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)溶于乙醇的特性,使其作为碱催化剂,结果釜式反应中原料在4小时内就实现了完全转化——比K2CO3催化的环化反应快得多(详细说明见附录A. S2.4)。在间歇釜式条件的基础上,单步的DBU催化环化反应首先在连续流过程中进行了试验。允化合物9的乙醇溶液、FAA和DBU的乙醇溶液同时进入反应器IV,反应器IV通过BPR加压,以防止溶液沸腾汽化(图6)。
首先,在85°C和5 bar背压条件下进行的流动反应的停留时间为20分钟,结果显示转化率较低(<10%)。当作者将反应温度提高到120°C时,转化率上升到75%。当进一步提高反应温度时需要增加背压,以保持乙醇溶剂为液体状态。经过进一步优化,在140°C和10 bar背压条件下,连续流反应的停留时间为30分钟,转化率良好(表5)。由此,连续流技术在处理低沸点溶剂中的高温反应的能力得到了充分证实和应用。

图6. 连续流合成化合物5,P15-P19为泵
表5. 在反应器IV中连续流动合成5的优化。

FP15:P15泵的流量
a通过GC/MS峰面积百分比确定转化率
为了连续流动合成化合物5,本文接下来准备从起始材料6开始,通过四步连续工艺制备吡咯[2,1-f][1,2,4]三嗪-4-胺(5)。通过将反应与分离单元整合,进一步实现全连续流工艺流程。RE-1的流出物料与FAA和DBU的乙醇溶液一起流入T型微混合器中,所得的混合物通过反应器IV进行反应(图7)。反应器IV的产物与饱和NH4Cl水溶液一起流入反应器m-CSTR-6(图7)。然后通过连续萃取分离设备ACE-2用乙酸乙酯进行萃取,以便将产物5连续提取到有机相中。有机相随后被泵入RE-2,通过减压浓缩后,快速去除溶剂(图7)。最终优化后的4步连续流过程的总停留时间为74min,化合物5 的分离收率为27.7%。
 图7. 连续流动合成4的示意图。P20和P21是泵。
图7. 连续流动合成4的示意图。P20和P21是泵。
最后一个反应过程(第5步)是吡咯[2,1-f][1,2,4]三嗪-4-胺(5)进行溴化反应,生成目标化合物7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)。在作者的前期研究中使用了1,3-二溴-5,5-二甲基海因(DBDMH)作为溴化剂进行了釜式溴化反应(详细说明见附录A. S2.5)。实验发现转化率和选择性的结果均较差(附录A. S1)。如果底物完全转化,则会存在过度溴化的现象,否则,化合物4的转化率较低(68%–64%),区域选择性也不高(63%–84%)。
接着,作者采用NBS作为溴化剂进行了实验。釜式实验结果表明,当反应温度为- 40°C时,用NBS进行溴化可获得良好的转化率和区域选择性,当反应温度将至- 78°C时, 转化率降至81%−78°C(附录A. S2)。在此条件下,我们使用纯化后的化合物5进行单步连续流溴化反应。在与釜式工艺相同的条件下,在温度−40°C时,作者观察到5完全转化,连续反应的停留时间为5分钟(表6)。
表6. V反应器中连续流动合成4的优化实验

FP20:P20泵的流量;FP21:P21泵的流速。
a转化率由GC/MS峰面积百分比确定。
b选择性由GC/MS峰面积百分比确定。
最后,本文考察了五步全连续流程中从原料6合成最终产物4的过程。作者将第5步连续过程集合到之前的四个步骤,RE-2的流出物料与NBS(1.05当量)的DMF溶液一起流入T型微混合器,然后将混合物泵入反应器V中。最后收集并纯化反应器V的产物。总体上,五步连续流过程生产7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)的分离收率为14.1%,总停留时间为79分钟,生产量为2.96 g·h−1。
连续流合成有两个显著的优点。首先,与传统的间歇釜式方法相比,连续流反应的时间显著缩短(表7)。在本研究中,间歇法的总反应时间超过19小时,而连续流的总反应时间仅为51分钟。由于间歇法中涉及的准备程序十分耗时,釜式合成中消耗的总时间(即反应时间加上反应后处理的时间)超过26.5小时(相应的处理过程总共至少需要7.5小时)。相比之下,连续流工艺中的总操作时间仅为79分钟,因此保障了目标化合物7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)的高效率生产。其次,与间歇釜式法相比,在连续流动条件下,生产的的安全性显著提高。
表7. 间歇釜式法和连续流动法的比较

a不包含后续处理时间
【结论】
1. 本文开发了一种五步全连续流动合成抗病毒药物瑞德西韦的核碱基单元7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)的工艺流程,起始原料为廉价且广泛可得的吡咯(6)。
2. 在连续流反应最优条件下,目标化合物4的分离产率为14.1%,连续流过程的总停留时间为79min,生产量为2.96g·h−1。
3. 该连续过程的总停留时间明显少于间歇釜式过程中消耗的总时间(>26.5小时)。
4. 本文的连续流合成过程涉及到危险和不稳定的中间体放热反应、液-液两相氧化反应和低温反应,这些反应过程均在连续流技术中得到了有效调控。
5. 多个后续处理过程,包括连续过滤、液-液分离、液-液萃取和减压蒸发等,可以通过专用的连续流动设备和装置被完全集合到连续反应的工艺序列中。
6. 将后续处理和多个化学反应过程整合到一个整体的连续流动系统中,可以避免费时费力的中间体分离和纯化过程,从而以最少的资源和能源消耗以及最少的废物排放实现快速高效的连续生产制造。
7. 本文的研究为下一代瑞德西韦药物合成方案的快速开发和可放大生产提供了连续流技术方面的参考。
一正科技简介:
作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。
公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。
公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
更多![]()

拓宽实习渠道,深耕校企合作 | 深圳职业技术学院学生参观一正科技实习基地
厂商
2022.11.17
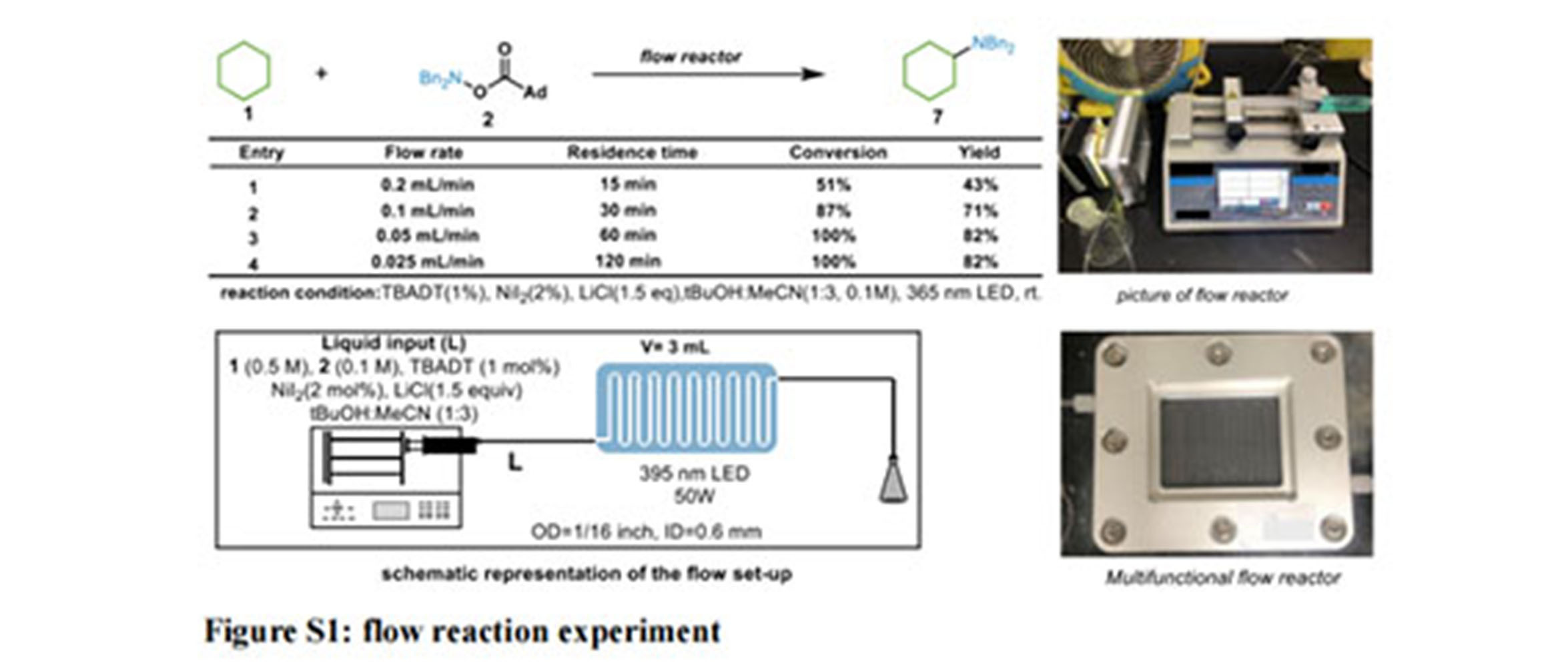
一正科技PER-10连续流光电化学反应器应用 【南京大学ACS Catalysis案例】光氧化还原活
新品
2022.09.15
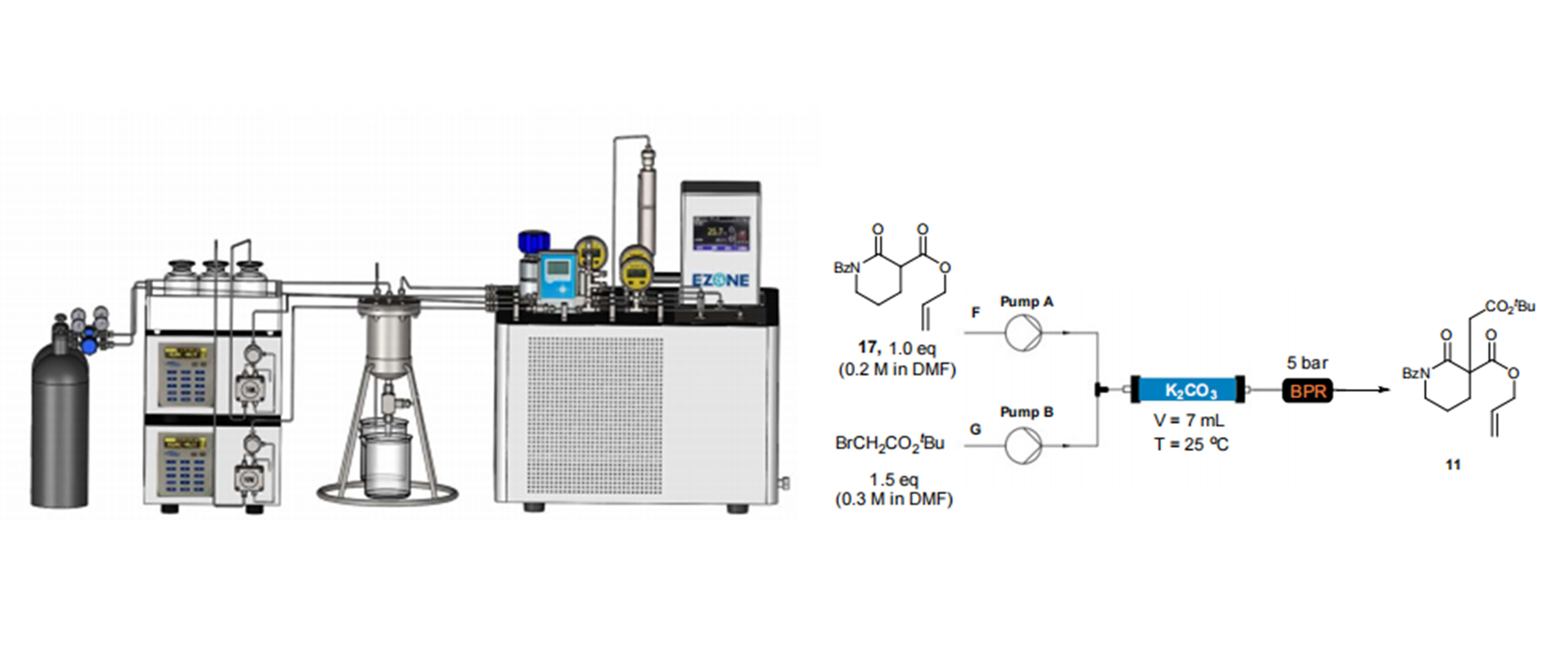
MF200微通道固定床应用案例【四川大学连续流案例]通过Pd催化的脱羧不对称烯丙基烷基化策略
新品
2022.09.15

中国化学会手性中国2021(2022)学术研讨会
新品
2022.08.23



公司名称: 深圳市一正科技有限公司
公司地址: 深圳市宝安区飞扬科技园A栋510 联系人: 李小姐 邮编: 518000 联系电话: 400-860-5168转1639
仪器信息网APP

展位手机站