
 关注
关注
已关注

![]() 已认证
已认证
粉丝量 0
400-860-5168转1639
仪器信息网认证电话,请放心拨打

拓宽实习渠道,深耕校企合作 | 深圳职业技术学院学生参观一正科技实习基地
2022年11月14日,深圳职业技术学院组织2021级精细化工专业学生到一正科技进行实习基地参观。期间,我司运营经理李嫣然对公司业务及实习岗位,待遇进行了详细介绍。深圳市一正科技有限公司成立于2006年,是深圳市高新技术企业、国家高新技术企业和深圳市高新技术产业协会会员单位。主营连续流微反应器产品及实验室自动检测化学仪器,为客户提供专业的连续流工艺整体解决方案和实验室系统解决方案。公司具有十几年的国外产品代理经验,也是国外众多知名品牌在中国的总代理和技术服务中心。近年由贸易型公司向研发生产型公司过渡。公司对各岗位人才求贤若渴,员工素质不断提高,目前,30%以上具有硕士及以上学历。经过不断的创新与累积,2015年公司通过了ISO 9001认证,具有Ezone自主品牌,取得了30余项实用新型专利及10余项发明专利,开发了微通道固定床反应器、连续流光电催化反应器等装置,在市场上得到良好的反馈,用户遍布高校研究院和药企、精细化工企业。2020年还参与多项深圳市科技项目,具有雄厚的技术实力。关于实习/兼职,目前可提供装配工程师、测试工程师、研发助理、商务/市场文员、销售助理等岗位,有系统的新员工入职培训,包括产品知识及技能培训等,专人帮扶指导,帮助同学们将所学理论知识融会贯通应用于实际工作,为以后就业求职打好基础。另外公司还提供优越的福利待遇:如5天7小时工作制,六险一金,法定节假日、带薪年假、超长福利年假;丰厚年终奖、出差补贴、通讯补贴、交通补贴;生日礼金/礼品、传统节日礼金/礼品、持续供应的零食点心、咖啡、茶叶等;每月都有的健身/娱乐活动、省内外旅游、出国游等。同学们对所提供的岗位及待遇非常感兴趣,会后与我司HR保持紧密的联系。我司也会努力回馈社会,提供更多的就业岗位及机会。【一正科技简介】一正科技成立于 2006 年,作为深圳市高新技术企业、国家高新技术企业和深圳市高新技术产业协会会员单位,2015 年公司通过了 ISO 9001 认证,具有 Ezone 自主品牌。一正科技注重自有知识产品的申请和保护,已取得了 10 项实用新型专利及 4 项发明专利。开发了微通道固定床反应器、连续流光电催化反应器等装置并在市场上得到良好的反馈,用户遍布高校研究院和药企、精细化工企业。 作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。 公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。 公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403
企业动态
2022.11.17
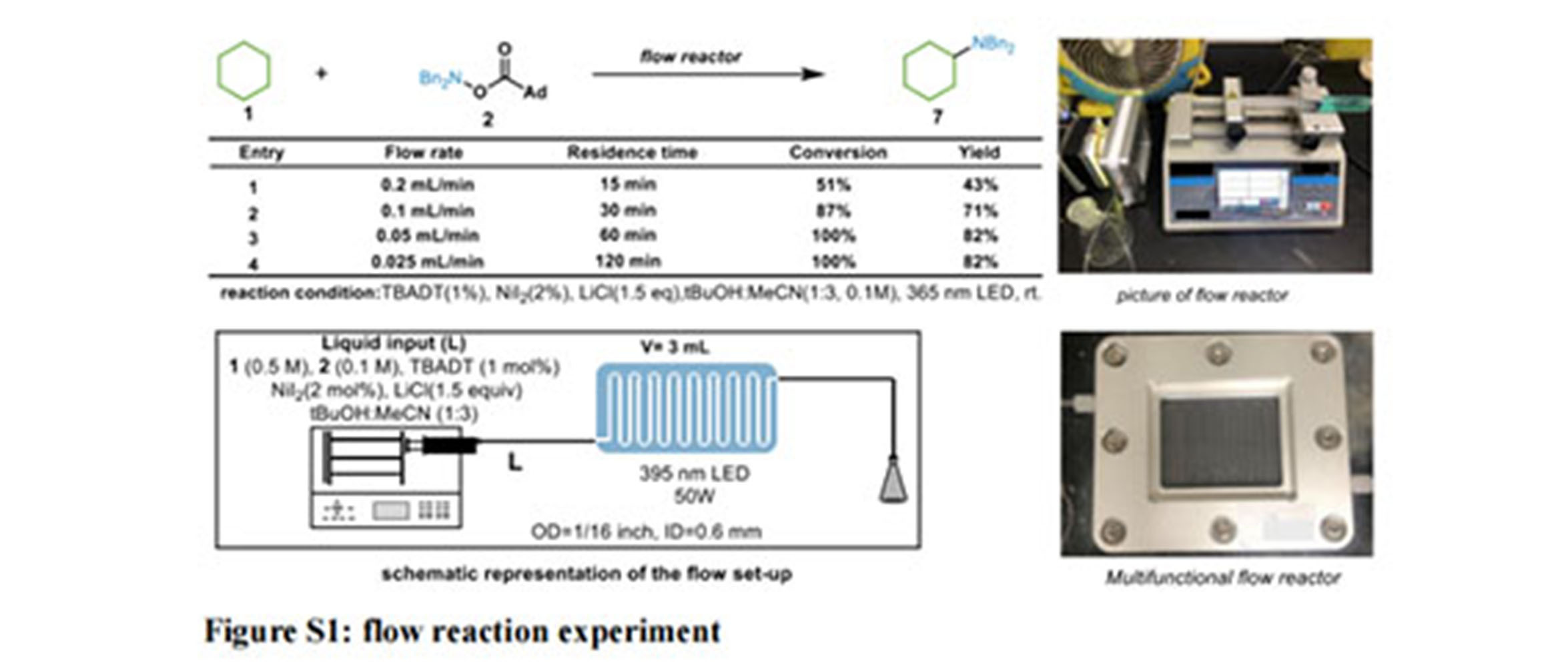
一正科技PER-10连续流光电化学反应器应用 【南京大学ACS Catalysis案例】光氧化还原活
【背景介绍】轻质烷烃是自然界中最丰富的资源,开发简便的方法实现烷烃转化为高附加值产品具有重要意义。然而,由非活化的烷烃来直接构建脂肪胺的研究案例却很少。南京大学化学院潘毅团队王毅课题组通过光氧化还原活性钨和镍接力催化,将轻质烷烃与胺/酰胺亲电试剂快速合成C(sp3)-N 键,同时结合连续流光电化学反应器实现了轻质烷烃的直接胺化。在过去的十年中,将烷烃转化为高价值化学品一直是有机合成研究热点。其中,C-N键的构建是主要任务之一。脂肪族胺广泛存在于天然产物、治疗药物、农用化学品和材料科学中。基于此,一些向烷烃分子中引入氮原子的方法已经被报道。目前,轻质烷烃的胺化主要有两种策略。首先是利用含有不饱和键的胺试剂(如偶氮、硝基等)与烷基自由基反应,通过自由基捕获实现C-N键的构建。另外一种是通过过渡金属催化的C(sp3)-H键胺化反应,主要经历金属亚胺中间体。然而,通过高价镍复合物中间体还原消除实现C(sp3)-N 键的构建还未见报道。近期,南京大学化学院潘毅团队王毅课题组报道了一种简便高效的C-N键的合成方法。光激发下,通过多聚坞酸盐选择性的攫取烷烃的C-H键,产生烷基自由基。随后产生烷基镍(I)物种。镍(I)物种与亲电胺试剂氧化加成得到关键中间体C-Ni(III)-N。通过还原消除得到目标产物。该研究结果发表在 ACS Catal. 2022, DOI:10.1021/acscatal.2c03456。首先,作者通过对添加剂,镍催化剂,光催化剂及溶剂得优化后,确定了多聚坞酸盐、无水碘化镍,氯化锂和叔丁醇乙腈混合溶剂的条件下具有较好的反应性。在最优的反应条件下,作者试验了各种非活化的烷烃底物范围。无论是烷烃的胺化还是酰胺化,对于环状烷烃都具有较好的反应性,同时,具有较好的位置选择性和官能团兼容性。对于酰胺化反应,同时适用于活化位点的胺化,例如烯丙基位,苄基位。随后,作者又试验了亲电胺试剂的底物适用范围。该反应体系同样适用于各种基团取代的亲电胺试剂。具有吸电子和给电子取代基的胺亲电子试剂在反应中是可以兼容的,甲基(23)、卤素(24、28、30) 、芳基(25)、甲氧基(27)、甲磺酸酯(29)和三氟甲基(31)等官能团在C-N 交叉键偶联反应过程中保持完整,杂环底物也以中等产率得到产物(32)。具有不同取代基的二恶唑酮(吸电子和供电子取代基)在反应中是可以兼容的,杂环(呋喃)取代的亲电试剂也能得到相应的目标产物。为了进一步扩展反应的可行性,天然产物和药物分子链接的分子在标准条件下可以以中等收率得到酰胺化产物。考虑到原子经济性问题,作者通过对反应副产物的分离,能以91%的回收率得到金刚烷甲酸。值得注意的是金刚烷甲酸是合成亲电胺试剂的起始原料。从表观来看,整个反应原子利用率极高。对于酰胺化反应来说,唯一的副产物是二氧化碳。所以综上来看,这种方法简洁高效,原子利用率较高。为了验证该反应工业放大的可行性,作者采用PER-10连续流光电化学反应器(深圳市一正科技有限公司)探究了实验室条件下的流动相反应过程。当反应液以0.2 mL/min的流速流动时,反应物的实际停留时间为15 min,能以43%的产率得到目标化合物。PER-10连续流光电化学反应器(深圳市一正科技有限公司)当降低反应液流速,以0.1 mL/min的流速时,相应的停留时间为30 min,以71%的产率得到目标产物。亲电胺试剂的转化率为87%。继续降低流速,当以0.05 mL/min的流速时,亲电胺试剂的转化率达到了100%,目标产物的产率为82%,相应的产物生成率为0.25 mmol/h。这为后续的工业放大研究提供了初步的探究。PER-10连续流光电化学反应器(深圳市一正科技有限公司)【结 论】随后,作者做了一系列机理验证得出以下结论:1. 通过自由基抑制剂证明反应可能经历自由基过程2. 光的开关实验证明了光照对于反应是必需的3. 通过KIE实验以及各组分的反应动力学分析,验证了催化剂的再生是限制反应速率的主要因素4. 在光照下,激发态光催化剂从烷烃中提取氢原子得到还原态光催化剂和烷基自由基R·5. 被光催化剂还原的Ni(I)物种得到Ni(0)物质,它捕获烷基自由基得到Ni(I)-烷基物种6. 二恶唑酮与Ni(I)-烷基物种的配位形成 Ni 配合物,其转化为亲电金属亚胺物种,并释放分子CO27. 氮烯插入得到Ni-酰胺复合物 E,它很容易质子化得到产物和Ni(I)并用于下一个循环相关成果近期在线发表于ACS catalysis。详细内容见:Photoexcited Direct Amination/Amidation of Inert C(sp3)?H Bonds via Tungsten?Nickel Catalytic Relay. Doi: 10.1021/acscatal.2c03456案例原文:/include/upload/kind/file/20220914/20220914140046_4478.pdf【一正科技简介】一正科技成立于 2006 年,作为深圳市高新技术企业、国家高新技术企业和深圳市高新技术产业协会会员单位,2015 年公司通过了 ISO 9001 认证,具有 Ezone 自主品牌。一正科技注重自有知识产品的申请和保护,已取得了 10 项实用新型专利及 4 项发明专利。开发了微通道固定床反应器、连续流光电催化反应器等装置并在市场上得到良好的反馈,用户遍布高校研究院和药企、精细化工企业。作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案
新品
2022.09.15
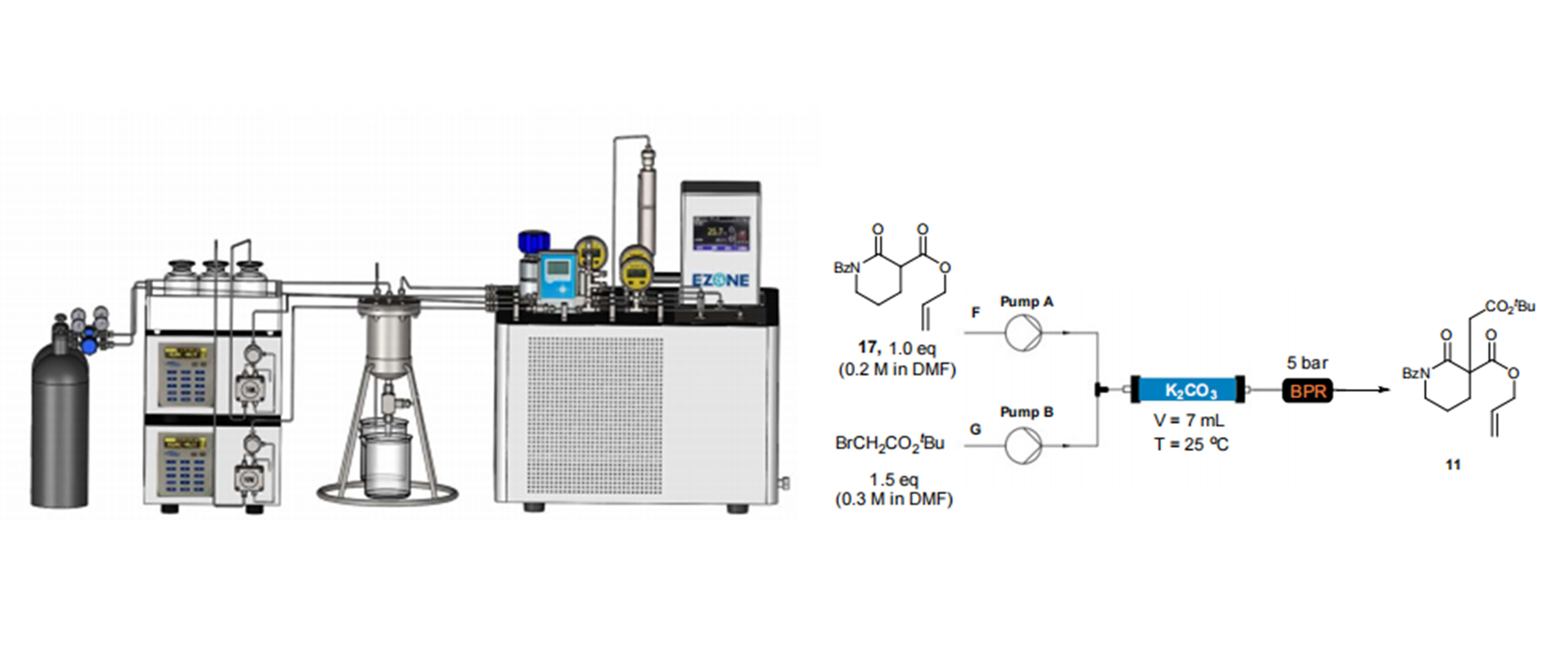
MF200微通道固定床应用案例【四川大学连续流案例]通过Pd催化的脱羧不对称烯丙基烷基化策略
【背景介绍】生物碱Schizozygine[1](例如,1-5,图1)几乎完全从东非单型灌木Schizozygia cofaeoides的树枝中分离出来,是eburnamine-vincamine[2]生物碱的家族成员,并显示出有用的生物学活性[3,4],如抗真菌、抗菌和抗疟原虫等特性以及其他药理特性。Schizozygia cofaeoides[5] (夹竹桃科)这种植物在肯尼亚传统医学中用于治疗皮肤和体表寄生虫疾病。(–)-Strempeliopine (4)和(–)-vallesamidine (5)从古巴物种 Strempeliopsis strempelioides K. Schum[6]中分离出来,与Schizozygine生物碱 (1-3) 相比,具有相反绝对构型。在结构上,(–)-strempeliopine (4) [7]体现了一个独特的类别,具有{六氢-乙醇吲哚[3,2,1-de]吡啶并[3,2,1-ij][1,5]萘啶环系}六环骨架,以及 (7R, 2S, 21R, 20R) 连续立体中心。图1. 具有代表性的Schizozygine生物碱由于其复杂的分子结构、潜在的生物活性以及低天然丰度,这些Schizozygine生物碱引起了合成界的广泛关注。针对这些生物碱的合成,合成界已经做了大量的工作[8],他们包括:1) Le Men课题组通过 (–)-tabersonine还原重排反应半合成了(+)-5[8a];2) Trojánek课题组用Le Men策略从(+)-18-亚甲基-乙烯基甲醛仿生合成(–)-4[8b];3) Heathcock通过八步反应从2-乙基环戊酮合成(±)-5,其特点是NBS介导的氨基内酰胺环化反应[8c];4) Padwa以分子内1,4偶极环加成反应和Heathcock的NBS诱导的环化反应为关键步骤合成(±)-4[8d];5) Okada以手性内酯为原料,以还原自由基环化为关键反应不对称合成(–)-5[8e];6) 秦勇课题组以光催化自由基级联方法合成(–)-4和(–)-5[8f,g];7) Anderson的不对称合成(+)-3和(+)-5涉及[1,4]-氢化物转移/Mannich环化反应[8h];8) Boger最近通过SmI2/BF3·OEt2诱导的脱芳跨环自由基环化反应不对称合成(–)-4,从而形成关键的C2-C21键[8i]。尽管取得了这些进展,但有效地获得具有高度立体化学控制的Schizozygine生物碱仍然是重要且艰巨的挑战。考虑到细微的结构差异对这一天然产物家族中的生物活性有巨大的影响,四川大学陈芬儿课题组发展了一种新的不对称全合成Schizozygine生物碱(–)-4的方法,这将为现有的半合成和全合成无法实现的灵活、深层次的结构修饰提供机会。本文报告了一种通用、高效的催化对映选择性全合成(–)-strempeliopine (4)的策略。文章发表在Chem. Commun., 2022, 58, 1402-1405(https://doi.org/10.1039/D1CC06278F)。【结果与讨论】本文对目标天然产物 (–)-strempeliopine (4) 的逆合成分析如方案1所示。设想4将从五环醛6通过跨环脱芳烃加成和Barton-McCombie反应获得。该已知方案将构建具有C2、C7、C21和C20连续立体中心的六环骨架,并为合成Schizozygine生物碱提供平台。N- 乙基吲哚-β-酰胺酯8的Bischler-Napieralski/内酰胺化级联可以完成五环亚胺7,并确保从受阻较小的 Si 面发生氢化物还原,从而得到6中的反式稠合八氢喹啉亚基。三环化合物8可以可通过已知色氨酸甲苯磺酸酯9和对映体纯的 N-苯甲酰基-β-酰胺酯10的偶联来获得。O- P- 众所周知,钯催化的内酰胺脱羧基不对称烯丙基烷基化反应(Pd-DAAA)已被证明是获得具有挑战性的具有四元立体中心的富含对映体的N-杂环的有力工具[9],有望发展成为立体选择性合成多种Schizozygine生物碱的通用方法的关键步骤。Q- R- 基于这一考虑,关键的底物10 反过来将由外消旋酰胺二酯11通过 Pd-DAAA 产生,以在 C20 位置建立全碳立体中心。二酯11可以很容易地从市售的2-哌啶酮12通过一系列常规官能团在化学中的转化来制备。本文的合成起始于以2-哌啶酮12应用连续流技术制备N-苯甲酰基-b-酰胺-二酯11[10]。最初,通过连续流技术,以苯甲酰氯和2-哌啶酮反应,得到内酰胺产物13。当等摩尔比的12和苯甲酰氯通过T形混合器在25 oC、10分钟保留时间和5 bar背压下流入PTFE线圈1(1 mL,0.8 mm i.d.)时,只有80%的转化率。当苯甲酰氯提高到1.2当量时,内酰胺产物13的分离收率为93%,底物12完全转化。接下来,在连续流动条件下,对化合物13中C3位置的酰化进行了大量的尝试,实验证明,对这一反应的优化难度比较大。将底物13的四氢呋喃溶液与LiHMDS 通过T 型混合器混合后泵入PTFE线圈 2 (2mL, 0.8 mm i.d.)。 再通过另一个T型混合器将混合物与氯甲酸烯丙酯14的THF溶液混合,并在-80 oC下以16分钟的保留时间和5 bar的背压流入PTFE线圈 3 (2 mL, 0.8 mm i.d.)。该反应进行得很顺利,但发生在底物12的C3位的酰化反应很少,区域选择性很差(17/18 = 3:4,难以分离,条目1,表1)。为了提高酰化反应的区域选择性,作者还研究了其他两种酰化试剂,当酰化试剂14被咪唑基甲酸烯丙酯15取代时,LC-MS未检测到O-酰化副产物18,在流动条件下(1.2当量LDA/THF,-80 oC,保留时间16分钟)生成所需的单酯17,产率为仅有32%(条目2)。令人高兴的是,使用氰基甲酸烯丙酯16作为酰化试剂在相同反应条件下以50%的分离产率得到了单酯17,没有副产物18(条目3)。 最终,通过优化了化合物13和化合物16的反应温度,从而以76%的收率得到了预期的酰化产物17,并且该反应的区域选择性得以保持(条目4)。最后,由于K2CO3微溶于DMF,因此选择了 MF-200固定床反应器(深圳市一正科技有限公司),以K2CO3作为填充材料。首先,将单酯17和溴乙酸叔丁酯的DMF 溶液用T型混合器混合,然后在25 oC和5 bar压力下将混合物注入到MF-200固定床反应器(7 mL内部体积)中,保留时间为10分钟。最终以87%的产率获得所需的二酯11。方案1. (–)-strempeliopine (4) 的逆合成分析表1. 通过连续流技术制备化合物11entryacylation reagentaconditionsselectivity 17/18 b.yield (%) c114LiHMDS (1.2 equiv.), THF, -80 oC, 16 min.3:4/215LDA (1.2 equiv.), THF, -80 oC, 16 min.> 99:132316 LDA (1.2 equiv.), THF, -80 oC, 16 min.> 99:150416LDA (1.2 equiv.), THF, -40 oC, 16 min.> 99:176[a] 所用酰化试剂为1.2当量。[b] 用LC-MS检测17/18的选择性。[c]分离收率。 Stoltz及其同事建立的内酰胺的Pd催化脱羧不对称烯丙基烷基化(Pd-DAAA)之后[11],为从11构建关键内酰胺 10 的关键转化奠定了基础。在80 °C[12],1,4-二氧六环为溶剂的条件下,研究了一系列与 Pd2dba3 结合的手性配体。观察到手性双膦配体(R)-BINAP L1[13a]为配体时,尽管反应产率高(92%,5% ee,条目1,表2),但对映选择性却很差。尽管 Trost 组通过使用2-二苯基膦基苯甲酸酯手性配体[13b]成功地完成了苯并稠合和非苯并稠合 δ-戊内酰胺的 Pd-DAAA反应,但(R, R)-Trost的配体L2和L3在这个反应中,效果不是很明显(条目2 和3)。底物10在用Pfalt配体 (R)-t-BuPHOX L4[13c]时,反应顺利地以94%的产率和51%的ee得到内酰胺产物10(条目4)。在改用(R)-(CF3)3-t-BuPHOX L5[13d]为配体后,反应的对映选择性显着提高(92%,88% ee,条目5)。受此结果的启发,继续使用L5进行条件探索,在1,4-二氧六环为溶剂,60°C下进行反应,得到91% ee的烯丙基化产物,产率为90%(条目6)。继续将温度降低到45 °C,得到相似的产率和稍高的对映选择性(90%,92% ee,条目7)。溶剂筛选表明,使用THF可以进一步增加对映诱选择性,可达94% ee,与用1,4-二氧六环时相近(条目8),而在MTBE或甲苯中的转化都顺利地提供了具有优异对映选择性的所需内酰胺,可达99% ee,且收率基本不变(条目9和10)。值得注意的是,这种转化有效地进行,并且以出色对映选择性实现克级反应(条目11),可以为后续的合成研究提供足够的底物。表2. 富含对映体的α-季胺内酰胺10的合成探索与优化研究entrylig.sol.temp. (°C)time (h)yield (%) bee (%) c1L1dioxane8049252L2dioxane80120103L3dioxane80120154L4dioxane80694515L5dioxane80692886L5dioxane601090917L5dioxane451590928L5THF451590949L5MTBE4515899910L5Toluene4515929911dL5Toluene45259099[a] 除非特别说明,反应均以0.1mmol 的底物11,5 mol %的Pd2dba3,12.5 mol %的配体在4 mL溶剂中进行。[b] 分离收率。[c] 以手性HPLC检测。[d] 1.2 g规模。在以Pd-DAAA反应得到关键的异构体10,随后作者将目标放到(–)-strempeliopine (4)的合成上。室温下,使用MeOH/H2O作混合溶剂,以氢氧化锂一水合物为碱,脱去苯甲酰基保护。可以以90%的收率得到脱保护的产物19,用于随后的缩合反应(方案2)。下步反应中,以NaH作为碱溶于甲苯中,在85 °C下,将底物19与色氨酸甲苯磺酸酯9反应,然后去除吲哚氮原子上的Ts基团,得到缩合产物8,两步反应总收率为45%[14,15],有了环化前体内酰胺8,就为构建关键五环核心的关键转化奠定了基础。令人高兴的是,底物8在POCl3的作用下发生Bischler-Napieralski/内酰胺化级联反应得到五环中间体,该中间体直接用LiClO4处理,得到稳定的亚胺中间体20[16]。以NaBH3CN作为还原试剂,还原亚胺20中的C=N双键,得到单一的具有C20/C21反式立体结构的非对映异构体21,两步反应总收率为65%。 通过NOE实验证实了21的相对立体化学,如方案2所示(橙色箭头)。因此,通过两步反应,能够制备具有令人满意的立体化学控制的关键五环骨架。作者推测是Re面会被烯丙基基团阻挡,还原性试剂优先从Si面接近,从而导致C20/C21的强反式选择性。方案2. 构建关键的具反式结构的五环内酰胺21根据合成计划,通过跨环去芳构化环加成来组装F环。为了将烯丙基转化为乙醛基,化合物21在臭氧条件下(O3,然后PPh3处理)以85%的产率得到醛6 (方案3)。受秦勇课题组的工作启发[8f],通过可调节的跨环结构构建 F 环,在 HMPA 和苯酚存在下,将底物6暴露于钐粉末和二碘乙烷中,可以以70%的产率得到六环仲醇22[17]。最后,通过Barton-McCombie自由基脱氧反应[18],以73%的总产率得到 (–)-strempeliopine (4),其分析数据与文献报道的数据相符合。方案3. 完成(–)-strempeliopine (4)的合成【结论】1. 本文的合成策略以Pd催化的脱羧不对称烯丙基烷基化(PD-DAAA)反应来构建具有挑战性的C20全碳四元中心为标志,通过串联反应Bischler-Napieralski/内酰胺/亚胺还原来构建关键的五环核心,具有高度的立体化学控制。最后通过改进的跨环去芳构化环加成反应构建了F环。2. 以商业易得的2-哌啶酮12为原料,经过13步反应实现了Schizozygine生物碱(–)-strempeliopine (4)的不对称全合成。3. 本文提出的合成策略可以为合成其他具有不同绝对构型的Schizozygine生物碱提供参考。【原文地址】Chem. Commun., 2022, 58, 1402-1405 (https://doi.org/10.1039/D1CC06278F)49661
新品
2022.09.15

中国化学会手性中国2021(2022)学术研讨会
本次会议由中国化学会手性化学专业委员会和清远中大创新药物研究中心共同主办,中山大学药学院承办,广东省化学学会、中山大学化学学院、广东省手性分子与药物发现重点实验室和广东省手性药物工程实验室协办,将于2022年08月26-28日在广东省广州市举行,08月25日报到。本次研讨会将邀请国内外学术和企业界的知名专家学者,共同展示手性科学与技术领域中最新进展和成果,为国内相关领域的科研技术人员提供一个良好的交流平台,欢迎手性化学及相关领域的科研、产业工作者积极参会。一、 会议主题:“手性科学让生活更美好”重点关注以下几个方面:1.手性合成新方法、新概念、新策略;2.手性放大的机制与规律;3.手性分子与生物大分子相互作用机理;4.手性物质的结构与功能关系;5.手性医药和农药生产的合成技术;6.手性大分子、手性超分子和手性材料。二、会议时间2022年08月25日(14:00—21:30)会议报到2022年08月26-28日会议2022年08月28日下午离会二、三、会议地点广州香岚官洲酒店(广州市海珠区广州国际生物岛星岛环南路1号)电话:020-89068888;020-80927526交通方案四、会议形式大会报告、获奖邀请报告、邀请报告、口头报告、研究生报告及墙报交流1)大会报告:(排名不分先后)周其林、涂永强、马大为、王剑波、张绪穆、刘鸣华、肖文精、刘维屏、刘冬生、胡文浩2)邀请报告:(排名不分先后)鲍红丽、段春迎、邓卫平、冯传良、顾振华、贾义霞、匡 华、李必杰、李原强、刘小华、刘心元、栾新军、梅天胜、苏成勇、孙建伟、唐智勇、徐森苗、杨海波、叶萌春、游书力、张万斌、赵晓丹、周佳海、朱 强3)口头报告个人申请,在提交会议论文时给予说明由学术委员讨论确定。4)研究生报告研究生报告为手性中国会议增加的新内容,旨在为研究生提供更好的交流平台和学习机会。请参与人员在提交会议论文时给予说明,最终报告名单将由学术委员讨论确定,望广大研究生踊跃参加。个人申请,在提交会议论文时给予说明由学术委员讨论确定。5)墙报展示个人申请,在提交会议论文时给予说明以便组委准备展板。墙报的规格84cm×119cm(宽×高,纵向,A0纸规格)。将在会议前通知墙报粘贴序号,请相关参会代表自带墙报,在志愿者协助下于指定地点张贴。五、会议日程六、会议主要事项1.会场屏幕显示为16:9格式。报告人提前到会场,并携带U盘将报告PPT提前拷贝到会场专用电脑;2. 本次大会的报告场次较多,大会报告时间不超过55分钟/人(含提问),邀请报告时间不超过25分钟/人(含提问),口头报告时间不超过10分钟/人(含提问),研究生报告时间不超过5分钟/人(不提问)。请各位报告人合理安排报告内容和时间。3. 准备展示墙报的代表完成注册后到3楼大宴会厅,在会务组的协助下张贴墙报(墙报的规格84cm×119cm宽×高,纵向,A0纸规格,需自行打印)。4. 本次会议报销需要的参会证明已统一放入会议手袋;扫描件参会证明、会议通知均在会议官网上可以自行下载,重要的注意事项也将在网上公布,敬请关注5. 具体的疫情防控方案详见会议官网;6. 请参会代表在会议期间全程佩戴代表证。七、联系方式会议网址:https://www.chemsoc.org.cn/meeting/CC2021/会议联系人:钱宇 qianyu5@mail.sysu.edu.cn 15221979602肖驰 xiaoch7@mail.sysu.edu.cn 15622394662【一正科技简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
新品
2022.08.23

化学加2022(杭州)中国医药CMC产业链技术交流暨企业家科学家高峰论坛
一:会议背景1、生物医药产业,近几年已经上升为全球各主要经济体,以及国内各发达省市的最高层战略。当下中国正处于生物医药发展的黄金时代,各医药研发企业正逐渐将研发重心从生物类似药研发聚焦到 First in Class生物创新药研发。随着企业研发能力提升,中国在一些前沿靶点领域超越欧美,完全有望实现技术输出。2020-2021年中国创新药 Licence-out在数量和金额上都呈现快速增长态势。纵观过去几十年中国多个行业的发展形势,我国生物医药创新还处于历史起步阶段,未来有巨大的可能性。不论是从科技、人才、高端仪器设备、市场需求,以及政策扶持方面,都将为生物医药的创新做稳步积累和积极推进作用。相信在不久的将来,一定将迎来国产创新药的成长期和爆发期。2、六年多来,化学加网举办过多种形式的线上线下精品活动,比如2017年的“庐山会谈”、2018年走进浙工大药学院、走进兰大化学院和南京药谷等大学园区、走进兰州白银经济区、2019年的“井冈山会谈”、化学加2021广州峰会、化学加2021成都峰会、化学加2022网络直播公益论坛(峰会盛况参见化学加视频号及化学加APP活动的相关报道)。众多药化产业企业家、科学家、投资家出席参会并做分享报告。本次化学加2022杭州峰会主要面向制药产业创新药研发企业(包括小分子大分子/ADC药物/双抗药物企业)、CXO企业、以及实验室建设/安全运营、绿色化学合成领域从业者。着重高层次前瞻技术研判、一线运营管理经验分享,药学/临床/制剂-CRO/CMO/CDMO服务创新合作对接,及部分优质项目产学研金融资源路演对接合作。届时深圳市一正科技有限公司副总经理钟明将分享报告主题:连续流工艺在精细化工及医药行业的应用,欢迎大家前来交流探讨。二:基本情况介绍会议时间:2022年8月15-16(星期一星期二)会议地点:杭州和达希尔顿逸林酒店(杭州经开区金沙大道600号)主办单位:化学加网承办单位:广州六摩尔生物医药有限公司协办单位:浙江工业大学药学院、上海月新智慧产业园、药智网支持媒体:药智网、化学加、药总汇、盖德视界会场设置:半天大会场 + 两大平行会场 + 一场回字型观众座谈 + 药智网半天专场(8月16日上午会场三)本次峰会规模约850人/1500人次(共两天,详见化学加APP/会议活动栏目介绍)会场一:包括新药研发、工艺研究、质量控制、临床前CRO、临床CRO、制剂改良、申报上市、投融资等主题会场二:医药CMO/CDMO工艺研究,绿色化学在医药产业的应用、药物筛选/AI应用/化学合成自动化;微通道连续流化学、实验室方案设计及运营管理、特色仪器设备应用于药化企业案例分享等主题三:报告嘉宾大会主席:林国强,中国科学院院士、中国科学院上海有机化学研究所研究员、上海中医药大学创新中药研究院院长、原中国科学院上海有机化学研究所所长报告题目:(待定)姜标,国际欧亚科学院院士,中科院上海有机化学研究所研究员,中国科学院曼谷创新合作中心主任,上海科技大学特聘教授,世界顶尖科学家协会执行理事长,中国化学会产学研促进委员会副主任报告题目:(待定)张彦涛,泰励生物科技(上海)有限公司联合创始人、董事长,曾担任美国礼来制药全球研发小分子设计与开发部门的首席科学家、礼来中国研发中心的创始董事总经理,及礼来对外合作研发亚太区副总裁报告题目:化学的挑战与化学家的责任四:会议议程会场一:包括新药研发、工艺研究、质量控制、临床前CRO、临床CRO、制剂改良、申报上市、投融资等主题1.上海美迪西生物医药股份有限公司创始人、董事长陈春麟博士:CMC研究和临床前研究的无缝对接关注点2.齐鲁制药(内蒙古)有限公司药物研究院院长、首席科学家黄科学博士:发酵来源的绿色农药原料药的现状与趋势3.深圳瑞思普利生物制药、珠海瑞思普利医药科技董事长兼首席科学家陈永奇博士:505b2产品的立项准备、研发关键和注册要点4.浙江大学药学院院长顾臻教授:血小板偶联药物5.海翔药业(股票代码:002099)研究院院长、上海海翔医药科技有限公司总经理赵富录博士:高效、低成本合成工艺开发策略与案例分析6.国家杰青、中科院上海药物研究所杨财广研究员:靶向“未靶蛋白质”抗肿瘤7.深圳湾实验室百瑞创新中心主任、重庆大学药学院贺耘教授:药物递送:药物开发“最后一公里”8.国家杰青、上海交通大学药学院院长张翱教授:二氟甲基化在药物发现中的应用9.暨南大学药学院教授、广州新济药业科技有限公司创始人吴传斌董事长:药物吸入制剂开发策略与难点分析10.浙江朗华制药有限公司总裁马建国博士:小分子药物基因杂质研究策略11.北京精金石知识产权代理有限公司专利分析师朱瑞:药品专利纠纷早期解决机制——药品专利链接制度12.上海博璞诺科技发展有限公司创始人、董事总经理朱文峰博士:基于有临床效用天然产物的创新药物13.长沙都正生物科技股份有限公司董事长欧阳冬生教授:基于药代动力学的改良型新药研发策略14.东莞暨南大学研究院院长、暨南大学南方药物经济学研究所所长蒋杰教授:创新药市场准入中的药物经济学研究15.医药政策研究专家、中国化学制药工业协会特约副会长兼政策法规专业委员会主任张自然博士:Biotech 商业化进展16.博济医药(股票代码:300404)副总经理、深圳博瑞医药科技有限公司总经理左联博士:CMC: 聚焦IND及I期临床阶段的小分子创新药物制剂开发研究17.清华大学药学院博士生导师胡泽平研究员:基于代谢组学和多组学技术的新药发现18.国际著名分子生物学家、生物化学&医药学专家、健艾仕生物医药科技(杭州)有限公司董事长傅新元教授:19.博腾股份(股票代码:300363)制剂研发副总经理赵建先生:外用制剂技术平台搭建和研发要点20.标新生物医药科技(上海)有限公司董事长兼首席运营官杨小宝博士: 分子胶和PROTAC交叉融合对于肿瘤和免疫药物的开发会场二:医药CMO/CDMO工艺研究,绿色化学在医药产业的应用、药物筛选/AI应用/化学合成自动化1.南方科技大学理学院副院长、南方科技大学坪山生物医药研究院院长、俄罗斯工程院外籍院士张绪穆教授:构建3S催化剂,发展绿色制药新技术2.华东师范大学化学与分子工程学院副院长高栓虎教授:天然产物合成及生物功能研究3.浙江工业大学药学院副院长、长三角绿色制药协同创新中心副主任、浙江省万人计划科技创新领军人才王鸿教授:天然药物新药研发与手性催化剂的构建4.上海弼领生物技术有限公司董事长兼CEO张富尧博士:新兴药物对合成工艺研究的挑战5.南方科技大学终身教授,国家“万人计划”专家,化学系副主任李闯创教授: 桥环天然药物全合成6.国家杰青、浙江大学求实特聘教授潘远江:现代质谱技术在药物分析中的应用7.北京深势科技有限公司创始人、CEO孙伟杰:AI+分子模拟技术赋能难成药靶标药物研发8. Elsevier生命科学解决方案中国区负责人俞靓:数据驱动下的药物研发工具——传统数据与AI预测应用及案例9.国家杰青、浙江大学化学系常务副系主任史炳锋教授:惰性碳氢键精准转化及应用10.国家杰青、华东师范大学姜雪峰教授:高价硫的点击化学11.北京师范大学化学学院党委书记卢忠林教授:荧光单元修饰大环多胺化合物作为非病毒基因载体的研究12.浙江工业大学药学院院长叶邦策教授:合成生物学与活菌诊疗13.中国科学院有机氟化学重点实验室董佳家研究员:一种可预测的,模块化的合成方法14.浙江大学化学系张玉红教授:15.Tillead Therapeutics Ltd. Co./苏州提领生物制药有限公司创始人、CEO赵红宇博士:16.药石科技(股票代码:300725)高级副总裁魏旭东博士:会场二:微反应技术、微通道连续流化学、实验室方案设计及运营管理案例分享等主题17.中科院大连化学物理研究所陈光文研究员(“微化工技术”研究组组长):微化学工程与技术18.国家杰青、清华大学研究生院副院长(挂职)、化学工程系徐建鸿教授:微反应技术及其在精细化工过程应用19.清华大学化工系助理研究员邓建博士:微反应技术与应用20.上海复旦大学手性分子与工程中心博士后姜梅芬:连续流动化学合成维生素B1中间体的工艺研究21.杭州精进科技有限公司董事长孔桂昌:连续化反应中实现精确稳定智能进料的技术研究与应用案例22.惠肤源生物技术(上海)有限公司CEO张辉博士:守护实验室安全,助力双碳达标——倚世在行动23.微反应器应用专家、浙江工业大学药学院夏春年副教授:微反应技术在智能医化的应用探索24.深圳市一正科技有限公司钟明副总经理:连续流工艺在精细化工及医药行业的应用……8月16日上午会场三:药智网活动专场8月16日下午会场三:回字形圆桌会谈(参会观众现场资源对接)五:峰会形式1、设半天大会场 + 两大平行会场,包含专题报告(30分钟/位)、圆桌对话(人均10分钟/位)、项目现场路演(设5分钟/位和10分钟/位两种),8月16日下午特别安排观众回字形圆桌会谈一场。2、参考化学加2021年10月成都峰会,本次大会从筹备到开幕,包括后续网上课堂,将通过化学加媒体矩阵全方位对外持续宣传。3、所有报名者,都有机会受邀作为分享嘉宾或者路演嘉宾。而且,如果您负责的“公司/项目/课题介绍PPT”同意授权公开,可提前发给化学加,部分将通过化学加微信公众号、化学加APP、化学加-网上课堂先行对外宣传展示。六:一正科技展台【一正科技简介】作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰ChemtrixB.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
新品
2022.08.04

关于召开“微化工技术研究与装备工程化应用研讨会” 的通知
微反应技术具有强传热和传质能力,实现化工过程的强化、微型化和绿色化;微化工设备具有高传递速率、可平行放大、安全性高、易于控制等优点,可实现化工过程的连续和高度集成的生产要求。因受疫情影响,原定在上海新国际博览中心召开的“(CTEF)2021 第十三届上海国际化工装备博览会( 2021 年 8 月 25 日-27 日)”与“(CTEF)2022 第十四届深圳国际化工装备博览会”合并于 2022 年 7 月28-30 日深圳会展中心(福田)举办。我单位将于 2022 年 7 月 27 日-29日在展会现场同期举办“微化工技术研究与装备工程化应用研讨会”,本次会议旨在展示和交流先进的微化工技术及设备应用,提升工业生产效能,创造拓展更多的机遇。届时将邀请业内专家,围绕微通道反应技术开发,先进设备的设计制造,和工程化应用等议题深入探讨。请各有关单位积极派员参加,现将有关事项通知如下:一、会议组织主办单位:中国化工企业管理协会承办单位:北京邦凯企业管理咨询有限公司广州振威国际展览有限公司二、时间地点时 间:2022 年 7 月 27 日-29 日(27 日全天报到)地 点:深圳市(地点确定直接通知报名者)三、会议费用会务费:2200 元/人(含会务费、资料费),每单位参会两人 1800 元/人。食宿统一安排,费用自理。四、拟邀出席专家及交流研讨内容本次会议将邀请高等科研院所及优秀企业等单位具有丰富理论造诣和实践经验的专家做主旨技术报告。届时深圳市一正科技有限公司副总经理钟明将分享报告主题:连续流工艺在精细化工及医药行业的应用,欢迎大家前来交流探讨。☆南京工业大学生物与制药工程学院副院长,教授、博导 方 正报告主题:微流场反应技术在材料及助剂开发与产业化中的应用☆河北工业大学化工学院教授 张月成报告主题:连续流微反应器在精细化学品合成中的应用☆上海交通大学化学化工学院教授 苏远海报告主题:复杂多相微反应过程强化基础及应用☆复旦大学手性分子催化与合成工程中心副教授 万 力报告主题:微化工技术在过程强化中的应用☆上海博瑞赛思化学科技有限公司创新技术经理 刘慧婷报告主题:模块化+数字化微反应器技术助力化学和制药工艺研发生产☆沈阳化工研究院化工新材料所所长助理、研究室总监 鄢冬茂报告主题:微通道反应技术在精细化工危险工艺中的应用☆清华大学化工系化学工程联合国家重点实验室助理研究员邓 建报告主题: 未来化学品制造模式☆上海惠和化德生物科技有限公司商务总监 马陈雷报告主题:基于微反应器的“Hybrid”工艺开发及工程化放大☆深圳市一正科技有限公司副总经理 钟 明报告主题:微通道连续流工艺在制药精细化工行业的应用☆北京日新远望科技发展有限公司教授级高工 张庆武报告主题:活性碳纤维在制药过程中的应用☆广州埃萌机电科技有限公司总经理 张千钦报告主题:浅谈连续流供料模块(泵与流量计等附件的)选型☆杭州精进科技有限公司总经理 孔桂昌报告主题:如何实现复杂条件下微化工及化学合成中的精确进料?☆郑州大学化工学院副教授 李文鹏报告主题:连续流反应与分离技术在精细化工中的应用☆中山致安化工科技有限公司总经理 欧志安报告主题:高通量工业微反技术创新及应用(其他相关专家报告正在预约中,敬请关注……)五、主要交流研讨内容(一)、微化工技术及微反应器的研究和应用现状;1、微化工技术研究与应用化进程;2、微反应器的研究与应用化进程;(二)、微化工技术与微反应器的行业应用与研究;医药行业领域1、微化工技术在医药行业的研究应用;2、微反应器在医药行业的研究应用;3、微通道在医药行业的应用研究;4、医药行业微通道反应验证及工艺开发等;5、医药行业微化工系统的放大和集成技术的研究;6、医药行业微反应工艺系统的优化设计研究与典型案例分析;农药行业领域1、微化工技术在农药行业的研究应用;2、微反应器在农药行业的研究应用;3、微通道在农药行业的应用研究;4、农药行业微通道反应验证及工艺开发等;5、农药行业微化工系统的放大和集成技术的研究;6、农药行业微反应工艺系统的优化设计研究与典型案例分析;染颜料行业领域1、微化工技术在染颜料行业的研究应用;2、微反应器在染颜料行业的研究应用;3、微通道在染颜料行业的应用研究;4、染颜料行业微通道反应验证及工艺开发等;5、染颜料行业微化工系统的放大和集成技术的研究;6、染颜料行业微反应工艺系统的优化设计研究与典型案例分析;纳米材料合成等领域1、微化工技术在纳米材料合成等领域的研究应用;2、微反应器在纳米材料合成等领域的研究应用;3、微通道在纳米材料合成等领域的应用研究;4、纳米材料合成等领域的微通道反应验证及工艺开发等;5、纳米材料合成等领域微化工系统的放大和集成技术的研究;6、纳米材料合成等领域微反应工艺系统优化设计与典型案例分析;其他精细化工领域;安全领域;(三)、微换热器研究与工艺优化中的验证及工艺开发应用;1、微换热器的研究现状和应用;2、微尺度下的传热特性;3、微换热器的结构优化研究;4、微换热器的可靠性与应用优点;5、微换热器的验证及工艺开发等;(四)、管式反应器应用的优缺点问题1、管式反应器的优势特点;2、管式反应器的典型反应;3、管式反应器在应用上的注意要点;4、工艺优化设计研究中管式反应器实践应用;(五)、在绿色化工过程中微化工技术的实际应用及典型案例;六、参会人员1、医药、医药中间体、农药、涂料、染颜料、香精香料等精细化工行业相关企业技术负责人;2、纳米材料合成等领域相关企业技术负责人;3、设备、技术供应商;4、政府、协会、检测机构、研究所及高等院校等。七、论文会刊征集本次大会将面向全国征集与主题相关的学术报告、论文、案例成果,印刷会刊(论文集)作为会议资料,请拟提交论文的人员在 7 月 20 日前将论文发至 zghg2012@126.com 信箱。要求论文字数不超过 5000 字,文件格式为 word 文档。八、联系方式组委会秘书处:电 话:13001080157(同微信)联 系 人:赵 蕊 电子邮箱:zghg2012@126.com九、附件十、公司简介深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
新品
2022.07.20
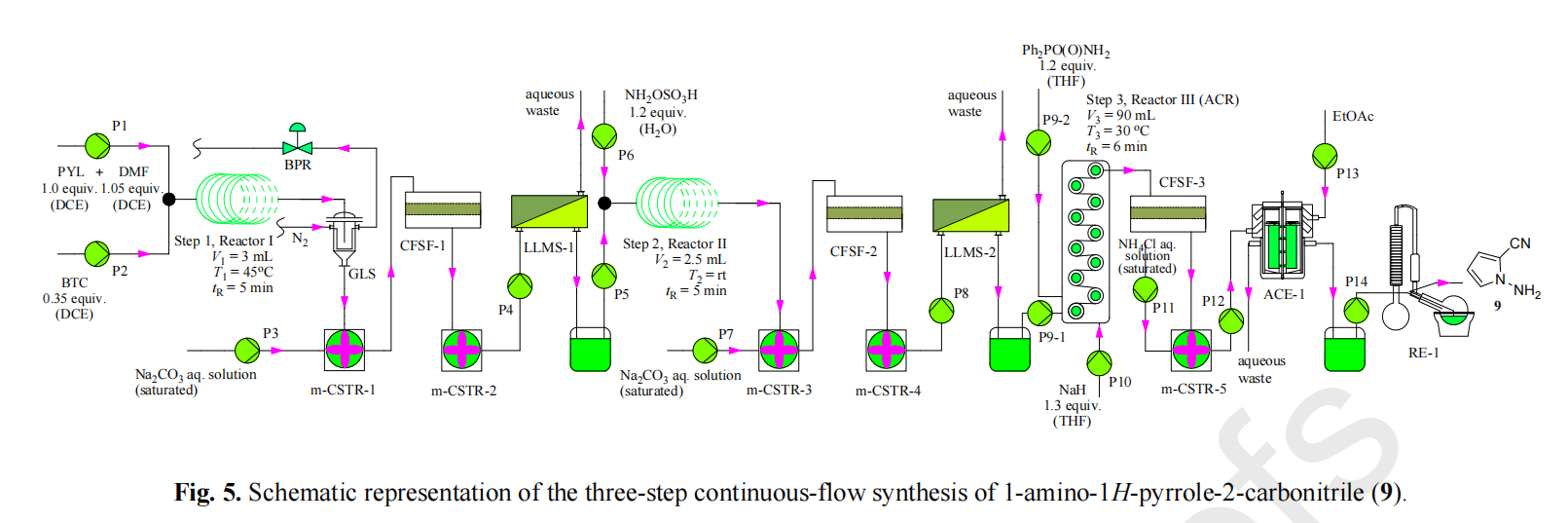
【复旦大学Engineering案例】采用ACR连续多级搅拌反应器、CSTR、固定床、连续膜分离装置等连续流合成瑞德西韦的核碱基单元
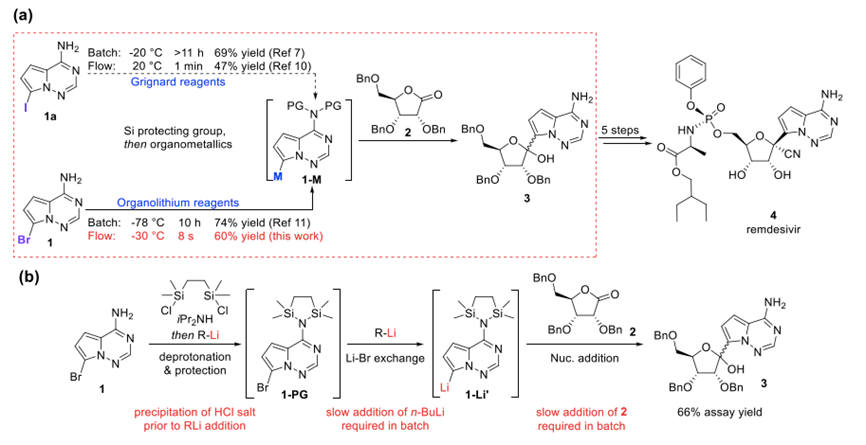
连续流合成瑞德西韦的核碱基单元【背景介绍】2019年严重急性呼吸综合征冠状病毒(SARS-CoV-2)引起的冠状肺炎(COVID-19)造成了全球毁灭性的公共卫生危机,瑞德西韦(Remdesivir)引起了世界的关注。这种磷酸核苷酸类似物被称为Veklury [1,2],由于专家对其有效性存在不同的观点[1,2,4],目前其仍在全球范围内进行单独或与其他药物联合使用的一系列临床试验,以期治疗感染冠状病毒的患者。在第一波新冠病毒大流行期间,瑞德西韦在美国和日本获准作为紧急药物使用,同时,在欧盟、新加坡、澳大利亚、韩国和加拿大获准有条件使用权限[1],允许住院的成人和儿童冠状疾病患者使用瑞德西韦治疗。 迄今为止,冠状病毒已在全世界造成约1.75亿人感染,380万人死亡[5]。更糟糕的是,全球确诊的病例数量仍在上升,一些更具传染性的变异株在世界各地引发了新的警报[6,7]。鉴于全球规模的病毒流行,以及生产瑞德西韦所需的活性药物成分(API)的巨大潜在需求,确保这种化合物的充足供应可能是一个关键问题[8]。尽管科研工作者已经在改进瑞德西韦的合成方法方面付出了很大的努力,但目前使用的间歇式批量生产仍然是一个漫长的、资源密集型的过程,必须按顺序完成,并且存在难以大规模操作、产量低的缺点[9]。 复杂的间歇过程似乎会影响在冠状病毒大流行等紧急情况下快速生产大量瑞德西韦的能力。此外,间歇式生产会导致高昂的成本,因此限制了这种药物的广泛使用[9]。因此,需要开发一种高效且可扩展的合成方案来制备瑞德西韦。通过逆合成分析(图1),瑞德西韦可以由三种不同的结构单元组成,包括核糖内酯单元(2)、具有立体生成磷中心的磷酰胺单元(3)和类似腺嘌呤的碱基单元(4;即7-卤代吡咯[2,1-f][1,2,4]三嗪-4-胺)[10,11]。 由于在后期进行有效地执行关键的C-糖基化步骤需要大量的核碱基单元(4),复旦大学手性分子工程中心的陈芬儿院士课题组在连续流系统中高效且经济地合成了核碱基单元(4),旨在开发一种独特的安全、高效、可放大的连续流系统,从而实现快速、可放大地制备瑞德西韦。该方案优化了涉及危险和不稳定中间体的反应,很好地控制放热反应,并且显著增强了液-液两相反应。此外,低温反应也得到了很好的调控。后续处理过程完全集合到了反应序列中,形成了一个整体的全连续流动系统,最大限度地提高了整体工艺效率。 图1. 瑞德西韦的逆合成分析 前期研究已经报道了几种合成路线来获得关键中间体吡咯[2,1-f][1,2,4]三嗪-4-胺(5)[12–16]。通过化合物5的简单溴化或碘化即可得到目标化合物4。在此基础上,本文描述了7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(X=Br时为4)的合成步骤。O'Connor等人[12]和Dixon等人[13]揭示了一种通过2,5-二甲氧基四氢呋喃与叔丁基氨基甲酸酯反应来提供NH-t-丁氧基羰基(BOC)保护的1-氨基吡咯的路线。随后与氯磺酰异氰酸酯和N,N-二甲基甲酰胺(DMF)进行氰化反应,得到NH-BOC保护的1-氨基-1H-吡咯-2-碳腈。BOC脱保护,然后在回流的乙醇中与乙酸甲脒(FAA)和磷酸钾环化,得到所需的中间体5 [12,13]。 Knapp等人[14]报道了一个类似的方案,其中5-二甲氧基四氢呋喃通过与2-氨基异吲哚-1,3-二酮或碳氮叔丁酯反应转化为1-氨基吡咯或NH-BOC保护的1-氨基吡咯。在甲酸中用NaOAc处理1-氨基吡咯,或在甲酸中用醋酐处理NH-BOC保护的1-氨基吡咯,均可产生N-(1H-吡咯-1-基)甲酰胺(N-(1H-pyrrol-1-yl)formamide)。随后与氰胺缩合得到N′-氰基-N-(1H-吡咯-1-基)甲酰胺;在刘易斯酸介导的环化后,得到化合物5 [14]。Patil等人[15]报道了一种两步法,其中吡咯-2-甲醛在水中用羟胺-O-磺酸(HOSA)和KOH处理,生成1-氨基-1H-吡咯-2-碳腈。 然后在碳酸钾存在下,在回流乙醇中用FAA处理色谱纯化后的1-氨基-1H 吡咯-2-碳腈,得到5[15]。然而,这些方法的实际应用受到起始原料的有限可用性和相对较高成本,以及分离相关中间体所需的快速柱层析的要求等限制。Paymode等人[16]最近报道了一种方法,首先通过使用磷酰氯(POCl3)和DMF的Vilsmeier–Haack反应对吡咯进行甲酰化。随后,通过与羟胺、醋酐和吡啶反应,将生成的吡咯-2-甲醛氧化转化为吡咯-2-碳腈。随后的N-氨基化和环合反应得到所需的5。这种方法的优点是使用了低廉的起始材料。 然而,POCl3的使用会导致含磷废水的产生,可能对环境产生严重的有害影响。同时,使用羟胺、醋酸酐和吡啶(3.5–5.0当量)进行氧化转化不仅增加了产品纯化的难度,还增加了制备5的总成本。此外,氯胺直接N-氨基化反应需要复杂的氯胺萃取和浓缩过程才可以提高产量,这将导致萃取溶剂和能源的大规模消耗。基于之前的研究,本文改进了合成方案,以无色谱纯化的方式从廉价且容易获得的吡咯中获得核碱基中间体4,如图2所示。首先通过Vilsmeier-Haack反应将吡咯(6)转化为吡咯-2-甲醛(7)。 本文没有使用传统的Vilsmeier–Haack试剂POCl3/DMF,而是使用双(三氯甲基)碳酸盐(BTC)和DMF来形成Vilsmeier盐。因此避免了含磷废水的产生。随后,在水中用较低成本的HOSA处理吡咯-2-甲醛(7),可以完成甲酰基官能团的氧化转化,从而获得吡咯-2-碳腈(8)。然后与O-(二苯基膦基)羟胺(DPPH)进行N-胺化,并与FAA环化,得到关键中间体吡咯[2,1-f][1,2,4]三嗪-4-胺(5)。最后,用N-溴代琥珀酰亚胺(NBS)溴化得到目标化合物7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4),该方案具有较高的区域选择性和产率。图2. 改进后合成化合物4路线从实用的角度来看,传统的间歇釜式方法在生产放大时可能会受到限制。首先,Vilsmeier–Haack和N-氨基化反应是高度放热的反应,并且都涉及危险化学品,在批量条件下很难控制。通常这种类型的反应都是在分批模式下进行的,即在低温下缓慢地向反应混合物中添加一种试剂,以防止由于釜式反应器中混合不足和散热缓慢而出现失控情况[17]。该操作适用于小规模的间歇过程,但由于比表面积降低可能需要过长的物料添加时间,不适合大规模生产。因此,大规模的釜式生产过程具有耗时、低效的缺点。 此外,由于快速反应和混合不足,如何保证釜式反应器中的温度均匀是一个重大挑战[18,19]。因此,可能会引发副反应和进一步的二次反应,导致产量损失和安全问题。其次,吡咯-2-甲醛(7)氧化转化为吡咯-2-碳三腈(8)是在不互溶的液-液两相混合物中进行的。传质速率在两相反应中起着重要的作用,在两相反应中,整体的反应速率受两相间活性物质转移速率的影响。因此,在传统间歇反应器中,氧化反应受到低效率的质量传递的影响,只有在有限的表面体积比(高达约2000 m2∙M−3)[20,21]中才可以实现,导致表观动力学受限。 此外,间歇式反应器中的两相流体动力学是基于反应器形状、尺寸、叶轮结构、搅拌速度等的一个众所周知的复杂函数。这使得反应器的放大变得十分困难,通常直到反应动力学和液-液流体动力学得到充分理解后才可以进行大规模的生产。第三,为确保形成目标化合物4的高选择性,溴化反应需要严格的低温条件(从−20到−78°C)。对于传统的间歇式反应器来说,有效且一致地维持低温是很困难的,因为当将间歇式反应器扩大到更大尺寸时,体积的增加速度远远快于外表面面积[22]。因此,间歇式反应器的低比表面积从根本上限制了生产的规模。 最后,间歇釜式过程由于是分步操作,涉及多步的反应序列、分离和纯化步骤[23]。通常,在每个合成反应完成后,需要将产物从反应混合物中进行分离并纯化,然后将获得的纯产物用于下一个反应。这种方法是劳动密集型、资源密集型、耗时且浪费资源[23]。最终,从起始原料到最终API产品(如瑞德西韦)可能需要长达12个月的时间,并且在不同的操作阶段需要大量的中间体库存[9]。 近年来,制药行业出现了向连续制造发展的趋势[25,26]。微反应器的使用在推动间歇式工艺向连续流工艺过渡方面发挥了重要的作用[27,28]。与传统的间歇式反应器相比,微反应器提供了独特的优势:小反应体积、快速混合和良好的质量和热传递,从而提高了反应性能和安全性,和对反应变量进行更精确的控制[29]。此外,与间歇过程相比,连续过程的生产放大要容易很多,可以通过对流动装置进行重复并联或放大反应体积来增加生产通量[29]。此外,通过使用连续流技术,可以将多个合成步骤集合成一个流线型生产体系,并且可以避免分离和中间体纯化的过程[30]。因此,使用最先进的连续流技术可以节省大量资源、空间、时间和能源。 本文计划通过采用连续流动技术克服间歇釜式合成的上述限制。本文目标是开发一种独特的安全、高效、可放大的连续流系统进行目标化合物4的合成。该方案优化了涉及危险和不稳定中间体的反应,很好地控制放热反应,并且显著增强了液-液两相反应。此外,低温反应也得到了很好的调控。后续处理过程完全集合到了反应序列中,形成了一个整体的全连续流动系统,最大限度地提高了整体工艺效率。 【实验方法】反应器I、反应器II、反应器IV和反应器V均是由内径(ID)为0.6 mm、外径(OD)为1.6 mm的全氟烷氧基(PFA)管构成。反应器III是一个连续搅拌反应设备(ACR;AM Technology,UK),其特点是一个安装在横向振动电机上的哈氏合金反应模块[31]。反应模块的中心流板包含反应室、互连通道和搅拌器。反应室由一系列约9.8 mL的反应单元组成,每个单元内有一个可自由移动的搅拌器。单个反应单元由长度为30 mm、宽度为4 mm的互连通道连接。当反应模块被振动电机振动时,搅拌器在单元中进行快速反转的横向运动[32]。 从原理上看,反应室模拟了毫升级连续搅拌釜式反应器(CSTR)的级联,以实现塞流特性。有关ACR的更多详细信息,可以参考其用户手册[31]。另外,本工作中使用最多的的是T型微型混合器来实现流体的混合。研究发现,对于混相流体,使用简单的T型微型混合器(步骤1、4和5)可以实现良好的混合。然而,当两股流体不相溶时,使用T型微混合器会导致反应流的不完全混合。 因此,在第2步中,采用了一种所谓的两相错流微混合器(CFMM)来混合不相容液体。此外,本文中还使用了其他几种类型的设备和装置用于将处理程序整合到反应序列中,包括气液分离器(GLS)、连续流固体过滤器(CFSF)、微型连续搅拌釜式反应器(m-CSTR)、液-液膜分离器(LLMS)、环形离心萃取器(ACE)和固定床反应器(FBR)。本文中使用的连续流设备和装置的详细说明见附录A.S3。 本文中使用注射器泵(Fusion 101、Fusion 200、Fusion 4000、Fusion 6000;Chemyx,美国)或高效液相色谱(HPLC)泵(SF1005A,Sanotac,中国)用于泵送溶液。反应料浆使用蠕动泵(德国Masterflex 77200-60型)泵送。背压调节器(BPR)购自Chemtrix(荷兰)。文中使用标准的1/4〃-28单向阀(美国IDEX Health&Science)防止逆流。流体连接采用标准的1/4〃-28螺纹接头,配有1/16〃盘管和卡套(中国Runzefluidsystem)。 【结果与讨论】第一步涉及吡咯(6)与DMF和BTC的Vilsmeier–Haack反应,从而生成吡咯-2-甲醛(7)。DMF和BTC之间的接触导致快速形成Vilsmeier复合物,该复合物具有热不稳定性,加热时可快速产生高温和高压[33,34]。这在大规模操作时容易导致安全问题[18,19]。因此,在间歇模式下,这种转化可以通过在0°C下缓慢地将BTC溶液加入吡咯和DMF的混合物中来实现[17]。添加完成后,将反应混合物加热至更高温度(45°C)进行进一步的反应。在间歇模式下完成毫摩尔级别的反应需要5个多小时(详细说明见附录A. S2.1)。 作者最初尝试在反应器I出口没有安装GLS的情况下进行流动反应,结果发现,6到7的转化率为95%,两股反应物(1.0当量吡咯,1.05当量DMF,0.35当量BTC,DCE)通过T型微混合器混合(图3中的步骤1),理论停留时间为30分钟(即反应器I的内部体积除以总流速)。然而,作者观察到流动反应器中会快速形成气体,使反应混合物的流速不稳定,导致实际停留时间无法控制。 为了解决这个问题,作者在反应器I之后连接了一个GLS,氮气(N2)通过加压气体入口进入GLS,并在其气体出口安装了一个可调节的背压阀(BPR)。在这种方法中,反应器I在BPR控制的设定压力下由N2稳定加压。然后将从GLS排出的反应混合物与进入的饱和Na2CO3水溶液一起流进m-CSTR-1(图3),可以促进中间产物盐的水解,从而获得甲酰化合物7。 图3. 两步连续流动合成吡咯-2-碳腈(8)的示意图。P1–P8为泵。PYL:吡咯;V:反应器的内部容积;T:反应器温度;tR:停留时间;rt:室温。 基于改进后的连续流系统,本文通过进一步的优化实验(表2)发现,35°C的较低温度和3 bar(1 bar=105 Pa)的背压可以实现在5分钟的停留时间内完成6到7的完全转化。 表2. 在反应器I中连续流动合成7的优化实验 FP1:P1泵的流量;FP2:P2泵的流量;P: 背压。a当停留时间不同时,摩尔比保持不变。b通过GC/MS峰面积百分比确定6的转化率。 完成生产化合物7的优化连续流方案之后,本文接下来计划将流程中的步骤2集合到步骤1中(图3)。随着反应器m-CSTR-1中形成固体盐,收集到的料浆被继续泵入到分离器CFSF-1中,用以去除可能堵塞下游反应器II的固体。在步骤2中,吡咯-2-碳醛(7)与HOSA发生氧化反应生成吡咯-2-碳腈(8)。在初步实验中,来自CFSF-1和HOSA水溶液的滤液通过两相CFMM直接流入反应器II。然而该方案并不令人满意,最佳结果化合物7只有79%的转化率,停留时间为30分钟。 CFSF-1滤液中水相的存在可能会影响后续步骤2中7的反应活性。因此,作者在CFSF-1之后加入了分离设备LLMS-1。滤液通过m-CSTR-2输送至LLMS-1,以连续的方式去除水相。从LLMS-1流出的有机相通过CFMM与HOSA水溶液混合,所得混合物进入反应器II。经过条件优化后,该优化方案在室温下以5分钟的停留时间将化合物7完全转化为8(表3)。 表3. 在反应器II中连续流动合成8的优化实验 FP5: P5泵的流量; FP6: P6泵的流量a当停留时间不同时,摩尔比保持不变。b 通过GC/MS峰面积百分比确定7的转化率。 接下来,反应器II的流出料与饱和Na2CO3水溶液一起流入到反应器m-CSTR-3中。由于固体盐是在m-CSTR-3中形成的,因此该料浆混合物随后通过CFSF-2以去除会堵塞LLMS-2的固体。LLMS-2可以快速分离两相混合物中的有机相。该优化后的两步反应-分离一体化连续流过程得到的化合物8分离收率为47.6%,处理量为3.02g·h−1。第3步反应涉及了吡咯-2-碳腈(8)的N-氨基化,从而获得1-氨基-1H-吡咯-2-碳腈(9)。该反应中有NaH的参与,尤其是在大规模生产条件下,会带来潜在的安全隐患。因此,釜式反应是通过将干燥THF中的底物8缓慢添加到NaH–THF混合物中,然后在THF中逐滴添加Ph2P(O)ONH2来实现的(详细说明见附录A. S2.3)。因此,釜式生产操作的效率和生产率都很低。由于NaH和Ph2P(O)ONH2不溶于四氢呋喃,并且产生了固体副产物,这些对将该反应转化为基于微通道的流动过程造成了阻碍。为了解决这个问题,本文使用了Coflore ACR反应器来促进料浆的流动。 考虑到要将ACR直接集合到之前两步流动序列的复杂性,作者首先考察了进行纯化吡咯-2-碳腈(8)的单步连续流动反应(图4)。将两股反应物(THF中1.3当量NaH、THF中1.2当量Ph2P(O)ONH2和1.0当量的化合物8)以特定的流速泵入ACR连续多级搅拌反应器中。作者发现化学计量比和停留时间对该连续流反应结果具有很大的影响,相关实验如表4所示。 图4. 单步连续流合成化合物9,P9–P14为泵 英国AM Technology 连续多级搅拌反应器ACR图片 表4. 在反应器III中连续流动合成9的优化实验 FP9:P9泵的流量;FP10:P10泵的流量。a因为形成的气体影响,这是测量后的实际停留时间。b 8的转化率由GC峰面积百分比确定。 结果表明,当NaH与Ph2P(O)ONH2的摩尔比小于1.17时,尽管停留时间相对较长,为16分钟,但化合物8最大转化率为50%。此外,当采用较高的摩尔比为1.63(NaH:Ph2P(O)ONH2)时,等量的化合物8仅在6分钟内就实现了完全转化。在最优连续流条件下,温度30℃、停留时间6min,最终获得了化合物9的分离产率为97%。值得注意的是,该反应在连续流中的反应时间仅为6分钟,而在间歇过程中则需要超过5小时(详细对比见附录A. S2.3)。此外,ACR连续多级搅拌反应器能够很容易地保持连续10小时以上的运行。因此,证明了使用ACR续多级搅拌反应器可以成功地进行混合料浆的连续流动过程。 随后,本文研究了从6合成9的三步全连续流过程。由于反应器II的流出物是DCE和水的液-液两相混合物,水的存在无疑会使NaH失活,从而阻碍步骤3的反应。因此,CFSF-2和LLMS-2依次被集合到反应器II之后(图5),以期对反应器II的输出物料进行连续过滤分离处理;然后,滤液流入LLMS-2分离出有机相。按照第3步的连续流优化方案,LLMS-2的有机相与THF中的Ph2P(O)ONH2(1.2当量)和THF中的NaH(1.3当量)一起被泵入ACR中,即可以实现9的三步全连续流动合成。 然后,通过CFSF-3进行连续过滤ACR流出的悬浮液。滤液与饱和NH4Cl水溶液一起流入m-CSTR-5中。然后通过ACE-1和EtOAc泵送出混合物,最终将粗产物9连续萃取获得有机相。ACE-1的有机相随后被泵入RE-1,经过减压浓缩快速去除溶剂。三步连续流合成化合物9的分离产率为44.3%,总停留时间为34min,产量为2.6g·h-1。 图5. 三步全连续流合成1-氨基-1H-吡咯-2-碳三腈(9)的示意图 在第4步反应中,1-氨基-1H-吡咯-2-碳三腈(9)在碱的催化下与FAA发生环化反应,得到吡咯[2,1-f][1,2,4]三嗪-4-胺(5)。前期的釜式环化反应是在85°C条件下,在乙醇中用纯化后的9与FAA和K2CO3反应进行的。该反应需要10小时才能达到完全转化(详细说明见附录A. S2.4)。由于K2CO3在乙醇中是以固体形式存在,作者尝试将K2CO3装填到FBR中,并泵送FAA溶液通过FBR以便将间歇过程转化为连续流。 然而,结果表明在这种模式下不能发生反应。然后作者利用1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)溶于乙醇的特性,使其作为碱催化剂,结果釜式反应中原料在4小时内就实现了完全转化——比K2CO3催化的环化反应快得多(详细说明见附录A. S2.4)。在间歇釜式条件的基础上,单步的DBU催化环化反应首先在连续流过程中进行了试验。允化合物9的乙醇溶液、FAA和DBU的乙醇溶液同时进入反应器IV,反应器IV通过BPR加压,以防止溶液沸腾汽化(图6)。 首先,在85°C和5 bar背压条件下进行的流动反应的停留时间为20分钟,结果显示转化率较低()。当作者将反应温度提高到120°C时,转化率上升到75%。当进一步提高反应温度时需要增加背压,以保持乙醇溶剂为液体状态。经过进一步优化,在140°C和10 bar背压条件下,连续流反应的停留时间为30分钟,转化率良好(表5)。由此,连续流技术在处理低沸点溶剂中的高温反应的能力得到了充分证实和应用。 图6. 连续流合成化合物5,P15-P19为泵表5. 在反应器IV中连续流动合成5的优化。 FP15:P15泵的流量 a通过GC/MS峰面积百分比确定转化率 为了连续流动合成化合物5,本文接下来准备从起始材料6开始,通过四步连续工艺制备吡咯[2,1-f][1,2,4]三嗪-4-胺(5)。通过将反应与分离单元整合,进一步实现全连续流工艺流程。RE-1的流出物料与FAA和DBU的乙醇溶液一起流入T型微混合器中,所得的混合物通过反应器IV进行反应(图7)。反应器IV的产物与饱和NH4Cl水溶液一起流入反应器m-CSTR-6(图7)。然后通过连续萃取分离设备ACE-2用乙酸乙酯进行萃取,以便将产物5连续提取到有机相中。有机相随后被泵入RE-2,通过减压浓缩后,快速去除溶剂(图7)。最终优化后的4步连续流过程的总停留时间为74min,化合物5 的分离收率为27.7%。 图7. 连续流动合成4的示意图。P20和P21是泵。 最后一个反应过程(第5步)是吡咯[2,1-f][1,2,4]三嗪-4-胺(5)进行溴化反应,生成目标化合物7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)。在作者的前期研究中使用了1,3-二溴-5,5-二甲基海因(DBDMH)作为溴化剂进行了釜式溴化反应(详细说明见附录A. S2.5)。实验发现转化率和选择性的结果均较差(附录A. S1)。如果底物完全转化,则会存在过度溴化的现象,否则,化合物4的转化率较低(68%–64%),区域选择性也不高(63%–84%)。 接着,作者采用NBS作为溴化剂进行了实验。釜式实验结果表明,当反应温度为- 40°C时,用NBS进行溴化可获得良好的转化率和区域选择性,当反应温度将至- 78°C时, 转化率降至81%−78°C(附录A. S2)。在此条件下,我们使用纯化后的化合物5进行单步连续流溴化反应。在与釜式工艺相同的条件下,在温度−40°C时,作者观察到5完全转化,连续反应的停留时间为5分钟(表6)。表6. V反应器中连续流动合成4的优化实验 FP20:P20泵的流量;FP21:P21泵的流速。a转化率由GC/MS峰面积百分比确定。b选择性由GC/MS峰面积百分比确定。 最后,本文考察了五步全连续流程中从原料6合成最终产物4的过程。作者将第5步连续过程集合到之前的四个步骤,RE-2的流出物料与NBS(1.05当量)的DMF溶液一起流入T型微混合器,然后将混合物泵入反应器V中。最后收集并纯化反应器V的产物。总体上,五步连续流过程生产7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)的分离收率为14.1%,总停留时间为79分钟,生产量为2.96 g·h−1。 连续流合成有两个显著的优点。首先,与传统的间歇釜式方法相比,连续流反应的时间显著缩短(表7)。在本研究中,间歇法的总反应时间超过19小时,而连续流的总反应时间仅为51分钟。由于间歇法中涉及的准备程序十分耗时,釜式合成中消耗的总时间(即反应时间加上反应后处理的时间)超过26.5小时(相应的处理过程总共至少需要7.5小时)。相比之下,连续流工艺中的总操作时间仅为79分钟,因此保障了目标化合物7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)的高效率生产。其次,与间歇釜式法相比,在连续流动条件下,生产的的安全性显著提高。表7. 间歇釜式法和连续流动法的比较a不包含后续处理时间【结论】1. 本文开发了一种五步全连续流动合成抗病毒药物瑞德西韦的核碱基单元7-溴吡咯[2,1-f][1,2,4]三嗪-4-胺(4)的工艺流程,起始原料为廉价且广泛可得的吡咯(6)。2. 在连续流反应最优条件下,目标化合物4的分离产率为14.1%,连续流过程的总停留时间为79min,生产量为2.96g·h−1。3. 该连续过程的总停留时间明显少于间歇釜式过程中消耗的总时间(>26.5小时)。4. 本文的连续流合成过程涉及到危险和不稳定的中间体放热反应、液-液两相氧化反应和低温反应,这些反应过程均在连续流技术中得到了有效调控。5. 多个后续处理过程,包括连续过滤、液-液分离、液-液萃取和减压蒸发等,可以通过专用的连续流动设备和装置被完全集合到连续反应的工艺序列中。6. 将后续处理和多个化学反应过程整合到一个整体的连续流动系统中,可以避免费时费力的中间体分离和纯化过程,从而以最少的资源和能源消耗以及最少的废物排放实现快速高效的连续生产制造。7. 本文的研究为下一代瑞德西韦药物合成方案的快速开发和可放大生产提供了连续流技术方面的参考。 一正科技简介:作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
新品
2022.06.06

【四川大学案例】采用一正科技微通道固定床反应器MF200、ACR连续多级搅拌反应器等实现全连续流不对称全合成四氢原小檗碱类天然生物碱
【背景介绍】近几十年来,连续流动合成技术在全合成中得到了广泛的应用。全连续流动合成在复杂天然药物分子的多步合成领域仍然备受关注。因此,研究天然产物的连续流动全合成是很有必要,且具有挑战性的。四川大学华西药学院陈芬儿院士课题组报道了一种全连续流动工艺技术,用于不对称全合成天然四氢原小檗碱类生物碱:(-)-四氢伪小檗碱、(-)-四氢伪黄连碱、(-)-四氢黄连碱和(-)-四氢小檗红碱。该技术流程包括四个化学转化和三个处理过程,在一个有序集成的连续流动平台中,无需任何中间纯化工艺。在32.5分钟的总停留时间内,四步全连续流动化学合成工艺的总收率和对映选择性分别高达50%和92%ee,生产效率为145 mg/h。然而,总体合成路线的评估不仅应强调新骨架结构和合成策略的创新,还应关注合成路线的原子经济性、[1]储存/时间经济性[2]和绿色可持续性。[3] 连续流合成作为一种新技术,与传统的间歇式合成方式相比,具有高效的传质/传热、易于放大、缩短反应时间和简洁的合成操作、更环保和更连续的优势,同时避免了中间体的纯化和分离过程。[4] 目前,有一些报告描述了生物活性天然产物[5-11]和活性药物成分(API)的连续流动全合成。[12,13]在这些工艺中,使用单步连续流或间歇和连续流操作相组合,也可以通过比单独间歇合成更高的产率和更短的反应时间得到复杂天然产物。尽管技术的进步使得使用连续流动系统可以合成相对复杂的天然产物。但这些连续流方法一般都会受到一些操作过程的限制,例如不可避免的离线纯化和分离,前后过程中溶剂的转换等。因此,开发包括在线处理过程在内的多步骤的全连续流动技术,必将成为天然产物合成领域的一个重大而艰巨的挑战。图1. 具有代表性的四氢原小檗碱生物碱的结构(-)-四氢伪小檗碱(1)、(-)-四氢伪黄连碱(2)、(-)-四氢黄连碱(3)和(-)-四氢小檗红碱(4)均具有复杂的多环二异喹啉骨架{四氢-6H-二苯并[a,g]喹啉锌},在C-14位置具有一个立体中心(图1),主要从小檗属和紫堇属植物中分离出来。[14] 该类型的许多化合物都表现出了很好的生物活性,包括抗精神病、[15a]抗炎、[15b]以及心血管疾病的治疗。[15c,15d]已知报道了两条四氢原小檗碱类生物碱1-4的不对称合成路线。[16] 2018年,Tong的团队开发了两条不对称路线,分别为不对称redox-A3反应(5步,2-7%总收率和77-99%ee)和基于对映体Noyori催化不对称转移氢化(6步,8-12%总收率和86-99%ee)得到了化合物1-3(方案1a)[16a]此外,Liu通过Noyori催化亚胺不对称转移加氢反应实现了4的13步全合成,总收率为18%,有很好地对映选择性(方案1b)。[16b]提高总收率和缩短反应时间对于这些天然生物碱的药物应用非常重要。本文中作者报道首个多步全连续流动工艺,用于化合物(-)-四氢伪小檗碱(1)、(-)-四氢伪黄连碱(2)、(-)-四氢黄连碱(3)和(-)-四氢小檗红碱(4)的全合成。方案1. 化合物1-4的合成路线总结方案2. 化合物1-4的逆合成分析【结果与讨论】本文首先在连续流条件下进行了二取代苯乙胺9和3,4-二取代氧基苯甲醛10a的还原胺化反应,在填充有10%Pd/C的微通道固定床反应器(型号:MF-200,厂家:深圳市一正科技有限公司)中制备仲胺7a。在60℃的温度下,背压为10 bar,使用甲醇作为溶剂进行连续流还原胺化反应,在10分钟的停留时间内,最终以84%的产率得到了仲胺7a(表1,条目1)。本文在相同的连续流条件下继续筛选了几种常见溶剂,如DCM、THF、甲苯和MTBE(表1,条目2-5)。结果发现,当使用MTBE作为溶剂时,产率较高为96%(表1,条目5)。接下来,本文研究了背压(从2.5 bar到15 bar)对反应结果的影响(表1,条目6-8)。研究发现,在5.0 bar背压下,产率增加到98%(表1,条目7)。为了提高生产效率,研究将停留时间从10分钟减少到2.5分钟。当停留时间减少到7.5分钟或5分钟时,可获得97%的产率(表1,条目8和条目9)。然而,在2.5分钟的停留时间时,产率降至90%(表1,条目10)。因此,该还原胺化反应的最佳条件是使用MTBE作为溶剂,背压为5 bar,停留时间为5分钟。表1. 连续流还原胺化的优化实验[a][a] 微通道固定床反应器中填充10%的Pd/C(5g)并用SiO2(2g)分散。 [b] 通过LC-MS测定产率。图2:微通道固定床反应器MF-200实现连续流还原胺化随后,在连续多级搅拌反应器(英国AM公司ACR反应器)中进行仲胺7a的连续成盐反应,该ACR连续多级搅拌反应器适用于反应系统中存在料浆的体系(表2)。[17] 从上一步获得的仲胺7a的MTBE溶液与盐酸的甲醇溶液按比例在ACR的第一个反应池中进行混合。本文考察了振荡频率和盐酸当量对反应产率的影响(表2,条目1-4)。当停留时间为10分钟,振荡频率5.0 Hz,使用4.0当量HCl时,反应可得到95%的较高产率(表2,条目4)。而当反应时间从10分钟减少到5分钟时,产率显著降低(表2,条目5)。反之,将反应时间延长至15分钟(表2,条目6),产率则略有提高。表2. 连续成盐反应的优化研究[a][a] 分离收率图3: 连续多级搅拌反应器中实现连续成盐反应在二氢原小檗碱5a的级联环化反应中,用二甲基缩醛8代替乙二醛,研究了四氢原小檗碱类生物碱四环核新骨架的构建。本文研发了一个集成的连续流技术平台,包括在线连续碱化(将pH值调整为10)、连续萃取和膜分离,可以连续有效地合成二氢原小檗碱5a。考虑到反应温度对二氢原小檗碱核心骨架的形成至关重要,文中采用了两个独立的PTFE盘管反应器,在60℃至120℃的温度范围考察温度对反应产率的影响。显然,随着温度从60℃提高到100℃,产率从8%提高到了68%(表3,条目1-3)。然而,由于二氢原小檗碱5a在高温环境下的不稳定性(表3,条目4),在120℃的温度下,所需产物的产率仅为12%。[18] 为了尽量减少二氢原小檗碱5a的分解,本文通过减小微反应器体积和提高流速来缩短反应停留时间(表3,条目5-7)。结果表明,在2.5分钟的总停留时间内,得到了70%的产率(表3,条目7)。之后,为了进一步提高产率,本文采用梯度温度策略,设置两个盘管反应器处于不同的温度条件(表3,条目8-11)。研究发现,当反应在80℃下平稳进行7.5分钟,在120℃下再进行2.5分钟时,产率达到了86%(表3,条目10)。在第一个盘管反应器中使用高温也会导致产品分解(表3条目11)。然而,当二甲基缩醛8的量减少到1.5当量(表3,条目12)时,产率明显降低。表3. 连续级联环化反应的优化实验[a] 通过LC-MS测定产率图4:微通道管式反应器实现连续级联环化反应(-)-四氢伪小檗碱(1)的全合成的最后一个步涉及连续流不对称铱催化的二氢原小檗碱5a的氢化反应。[19] 将从上一步获得的5a DCM溶液与铱催化剂和手性配体溶液在第一个T形混合器中进行混合,然后在第二个混合器中与氢气混合。研究发现含(S,S)-f-连萘的二茂铁配体是该催化不对称加氢反应的特殊手性配体。[11c]同时,本文选择DCM和AcOH的混合溶剂作为最佳反应溶剂。实验结果表明,增加压可以促进加氢反应的进行(表4,条目1-3)。由于连续流系统的压力上限,本文在40 bar的背压条件下进行了进一步的反应优化研究。当反应温度从20℃提高到50℃,反应时间从10分钟延长到30分钟时,反应产率和对映选择性略有提高(表4,条目3-8)。在80℃的条件下,停留时间为10分钟时,起始原料5a即可完全转化,获得(-)-四氢伪小檗碱(1)的分离产率适中,但由于温度较高,ee为85%(表4,条目8)。另外,停留时间的延长会导致反应产率明显降低(表4,条目9)。但是,如果将停留时间减少到7.5分钟(表4,条目10),则可以获得较高的产率和优异的对映选择性。表4. 连续铱催化不对称加氢反应的优化研究[a][a] 流动条件:Ir催化剂/配体/添加剂/底物=0.5:2:10:100,原料A溶解在DCM中,原料B溶解在DCM/AcOH(4:1,v/v)中。[b] 使用二氢原小檗碱5a时未经之前的环化纯化。[c] 从仲胺盐酸盐6a中分离出产率。[d] 通过手性HPLC分析确定对映体过量。[e] 反应体积为12.8毫升。[f]反应体积为19.2毫升。图5:不锈钢管式反应器中实现连续铱催化不对称加氢反应在四个单独的连续流过程中完成了(-)-四氢伪小檗碱(1)的全合成后,本文致力于连接这四个连续流过程开发一个完全连续的端到端过程,无需离线纯化过程或者重新优化反应条件(详见图S1)。通常,以高胡椒胺9和3,4-二甲氧基苯甲醛10a为起始原料,在固定床反应器中通过连续还原胺化引入四氢原小檗碱类生物碱的A/D环。然后,将获得的仲胺7a的MTBE溶液直接泵入到连续振荡反应器(ACR)进行成盐反应,从而获得仲胺盐酸盐6a的悬浮液料浆。为了实现从成盐到级联环化的顺利连接,本文采用了在线连续过滤装置来实现反应系统的溶剂交换。在减压浓缩设备中去除混合溶(MTBE/MeOH)后得到粗盐酸盐6a固体。然后泵入98%甲酸溶液中重新溶解盐酸盐6a,同时去除MTBE/MeOH溶剂的残留物,不需要经过干燥。随后,在两个PTFE盘管反应器中,通过Pictet-Spengler/Pomeranz-Fritsch级联环化反应,将6a的98%甲酸溶液与二甲基缩醛8的98%甲酸溶液进行混合,从而形成A/B/C/D 四环骨架。然后,将反应物料输送入包括在线连续碱化、连续萃取和液-液分离等单元的集成连续平台,以得到5a的DCM溶液,该溶液可直接用于后续不对称氢化反应,无需进一步的纯化。最后,将流出物料泵入1/8英寸的不锈钢盘管反应器中,通过连续的铱催化不对称氢化获得(-)-四氢伪小檗碱(1)。值得注意的是,这种新的全连续工艺以43%的总收率和91% ee得到了(-)-四氢伪小檗碱(1),总停留时间为32.5分钟,生产效率为133 mg/h。此外,本文还完成了其他相应的四氢原小檗碱类生物碱(-)-四氢伪黄连碱(2)、(-)-四氢黄连碱(3)和(-)-四氢小檗红碱(4)的多步全连续流的全合成,均得到了令人满意的产率和对映选择性和优越的生产能力(方案3)。总之,本文成功开发了适用于不同的底物的四氢原小檗碱类生物碱全合成的全连续流动体系。与传统的间歇釜式操作(方案3)相比,[20]这种多步骤全连续流合成工艺具有反应效率高、时间经济性好、合成路线可持续性高、高通量等优点。尤其,该方案使全合成的反应时间从100小时缩短到了32.5分钟,导致生产效率提高了300多倍(详见方案S1和S2)。此外,与已报道的四氢原小檗碱类生物碱1-4的合成路线相比,[16]这种新的多步骤全连续流动策略具有最短的合成步骤和最佳的总收率。值得注意的是,生产效率显著增加了近两个数量级(表5)。本文中所获得的(-)-1–(-)-4的化学结构均得到了核磁共振、红外光谱和高分辨质谱以及比旋度的证实(详见附录)。方案3. 连续流方法和间歇釜式方法合成四氢原小檗碱类生物碱1-4的比较[a][a] %ee由手性HPLC分析确定;从9为起始原料合成的纯化样品计算四氢原小檗碱的总收率表5. 四氢原小檗碱类生物碱1-4的不同合成策略结果对比。图6: Setup diagram of fully continuous flow (step 1-2)图7. Setup diagram of fully continuous flow (step 3-4)【结 论】1、本文开发了一种简洁的多步骤全连续工艺体系,适用于不同底物,可用于天然四氢原小檗碱类生物碱的全合成,包括(-)-四氢伪小檗碱(1)、(-)-四氢伪黄连碱(2)、(-)-四氢黄连碱(3)和(-)-四氢小檗红碱(4)的全合成。2、 该全连续流工艺共包含了四步化学反应过程和六种不同连续流设备中的三种在线处理过程,起始原料为廉价的仲胺盐酸盐和二甲基缩醛。反应过程包括合成仲胺的还原胺化反应、合成仲胺氢氯化物的成盐反应、形成二氢原小檗碱核心骨架的Pictet-Spengler/Pomeranz-Fritsch羟基烷基化/脱水级联反应,以及不对称铱催化氢化反应,最终生成四氢原小檗碱类生物碱。3、 该全连续流工艺的总收率和对映选择性分别高达50%和92%,总停留时间为32.5分钟,无需进行中间体的纯化处理,生产效率为145mg/h。4、 该研究不仅为四氢原小檗碱类生物碱的全合成提供了一种可扩展的连续流技术手段,而且为将来进一步探索其他相关衍生物提供了一个很好地参考平台。【原文地址】Chem. Eur. J. 2022, e202200700(https://doi.org/10.1002/chem.202200700)附件:/include/upload/kind/file/20220512/20220512173659_2712.docx【公司简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403
应用实例
2022.05.13

【荷兰格罗宁根大学案例】利用德国CINC连续离心萃取器实现两相生物催化连续反应
间歇生产是精细化学品制造的最新技术,因为使用的反应器针对不同的工艺表现出了多用性。然而,使用间歇反应器有一些巨大的缺陷:对于大批量生产,必须进行多次间歇运行,这常常导致不同批次间产品的质量和性能不稳定。此外,生产率往往低于专用的连续反应器,而且因为其劳动密集型的特点,导致维修成本非常高。然而,连续工艺对此则表现出了巨大的优势。此外,在小流量反应器中的连续生产对于使用或生产高毒性和/或爆炸性物质的反应非常有利。基于这一分析,许多科研小组已经开始研究过程强化的概念,其目的是发展小型反应器或将反应器与分离相结合。这一领域最突出的研究无疑是微反应器的使用。Poechlauer和他的同事们最近报道了使用微结构反应器进行吨级的Ritter反应。和田和吉田报道了微反应器中格里纳德交换反应的中试规模生产。还有学者研究了酶作为催化剂在微反应器中的应用。Ley和Baxendale发表了一系列论文,描述了微反应器中的串联反应,其中试剂以固定的形式存在。连续流反应器中的串联催化原理是一个非常有趣的问题。为了整体高效,每一级反应器中都要达到高转化率。然而,并没有多少反应其速度快到可以用在微反应器中,因为微反应器的停留时间通常只有几秒。此外,这些概念中的反应器并不总是容易放大到吨级。荷兰格罗宁根大学的Gerard N. Kraai等学者使用实验台规模的德国CINC连续离心萃取器系统研究了化学和生物催化反应,取得了重要的成果,并于2008年发表在《Angewandte Chemie》杂质上(Angew. Chem. Int. Ed. 2008, 47, 3905 –3908)。该装置是一种接触式连续离心萃取器(CCS),只有实验台规模大小,用于油水分离(如清理溢油)、连续萃取发酵产物(如青霉素和苯丙氨酸)以及在原子废物工业中用于提取和净化放射性废物。图1展示了CCS的结构示意图。该装置实质上是一台离心机。在转子外部与外壳体内部的小环形混合区引入了不互溶的液液两相。在这里,两相之间发生了非常高效且快速的混合,非常有利于两相催化反应。而后分散相被吸进离心机,在那里两相逐步但非常有效地分离,同时向上移动,之后通过不同的出口离开设备。 图1之前还没有报道使用CCS作为化学反应器。此离心机可以用于在间歇模式下连续地从蔗糖的酶转化过程中分离出聚合产物(右旋糖酐)。在液液两相(催化)反应中使用CCS具有潜在的吸引力。在这种情况中,环形区域作为反应器,离心机作为液液分离器。de Bellefon和Claus等学者报道了流动装置中的两相催化,但没有完整的相分离。Ryu和他的同事报道了一个微反应器中的两相Heck反应,其中含有钯催化剂的相是离子液体。在这种情况下,催化剂的分离和回收是完全结合的。通过连接一系列这样的设备,使用不同类型催化剂的串联催化在原则上是可能的,如图2所示。 图2作者测试了CCS对葵花油连续生产生物柴油的效率。该反应是典型的液液催化反应。在高温(60℃)下,以葵花油和6倍摩尔过量的甲醇为实验对象,使用碱性催化剂(NaOMe,1% w/w相对于葵花油)。CCS配备了加热套,以确保等温条件。葵花油被预热到60℃,并以12.6 mL/min的流速泵入CCS的一个入口,然后以3.1 mL/min的流速将MeOH中的NaOMe溶液泵入另一个入口。约40分钟后,体系达到稳定状态,含有部分残留葵花油的脂肪酸甲酯(FAME)以轻相形式析出,而重相为MeOH中的甘油溶液。调节不同的离心速度,在这些条件下FAME的收率最高可达到96%(图3)。 图3转速为30 Hz时转化率达到最大值。以更高速度旋转的离心机在分离时所增加的功率会导致发生反应的混合相体积减小(图4)。当离心机转速降低时,混合过程的效率降低,导致分散相液滴的平均尺寸增大,从而降低了传质速率和转化率。 图4利用所确定的最佳工艺条件,以61 kg/m3· min的产率生产生物柴油,比典型间歇法42 kg/m3· min的产率更高。此外,目前的工艺效率更高,因为没有单独的分离步骤,可以省去不同批次之间的反应器清洗步骤。接下来,作者研究了用CCS进行酶催化转化的潜力。大多数酶在水环境中功能最佳,因此是在CCS中测试的理想催化剂。由于酶很容易受到剪切力的破坏,因此采用低混合底板的CCS。该底板与离心机周围的保护筒连接,从而避免了进入的液体与旋转离心机直接接触。以油酸和1-丁醇为模型反应,研究了Rhizomucor miehei脂肪酶催化油酸与1-丁醇的酯化反应(Scheme 1)。 脂肪酶催化油酸和乙醇的反应已经为人所知,但作者发现用1-丁醇代替乙醇所得到的转化率要高得多。另外,作者还介绍了用青霉菌coryophilum的粗提取物在胶束体系中将丁醇和油酸酯化的方法。在间歇模式下,尽管存在大量过剩的水,这个反应仍然完全转化。据推测,该反应是由反应物的亲脂性引起的。在第1组系列实验中,使用了一种有机相,组成为庚烷中油酸(0.6 mol/L)和1-丁醇(0.9 mol/L)的混合物。水相由在pH=5.6的磷酸盐缓冲液中的R. miehei脂肪酶(1 g/L)溶液组成。作者首先研究了两相流量和离心机转速对转化率的影响(图5)。在这些条件下,作者发现转速为40 Hz,两相流量均为6 mL/min时达到了最高的稳态转化率(70%)。转化率显示了与每相流量相关的一个明显的最大值。在较低的流速下,CCS中以混合相为代价进行了更有效的相分离,其效果可与高转速相媲美。在较高的流速下,CCS中的停留时间过短,也会导致转化率降低。在这种特殊情况下,每相的最佳流速为6mL/min。与生物柴油的情况相似,离心机转速对油酸的转化率有着较大的影响,最优值为40 Hz。图5使用上述确定的最佳条件,在较高的酶负荷下(3.0而非1.0 g/L)进行脂肪酶催化酯化反应(图6)。大约2小时后,转化率变得相当稳定并在78~87%之间波动,平均82%,重复性良好。 图6在之前的实验中,酶溶液均为单次使用。为了提高酶的循环次数,作者进行了酶溶液连续循环和有机相部分循环的实验。有机相回收率为90%,油酸转化为油酸丁酯的转化率接近80%(图7),反应器在此模式下运行了13h,每克酶可产生486 g油酸丁酯。虽然随着时间的推移转化率会有所下降,但考虑到离心机的高速运转,可以认为酶在这段时间内保持了显著的稳定性。酶的稳定性仍然是未来发展的关键问题。图7中观察到的催化剂失活可能有许多不同的原因:最有可能的假设是,作为酶抑制剂的有机组分在水相中积累。这一假设目前正在求证中。 图7实验结论:Ø 在实验台规模大小的德国CINC连续离心萃取器中进行连续的化学和生物催化转化是可能的,也是非常有利的;Ø 即使在目前可以放在通风柜里的低成本设备中,也有可能在几天内生产100 kg的化学品;Ø CINC连续离心萃取器为已经成熟商业化的连续反应分离器,具备很好的规范性和通用性。随着CCS大规模投入市场,利用两相催化连续生产吨级精细化工产品的前景将变得十分广阔
应用实例
2022.04.11

【河北工业大学案例】采用荷兰Chemtrix玻璃微通道反应器Kiloflow光催化案例—2,6-二氯甲苯氧化溴化制备2,6-苄溴
【背景介绍】2,6-二氯苄溴是合成生物活性分子的重要中间体,如功能化[1,4]-噻嗪、4,6-二芳基嘧啶-2(1H)-酮和2-苄氧基苯甲酰胺(图1)。1-3 2,6-二氯苄溴通常是由2,6-二氯甲苯与Br2在自由基引发剂存在下或在光照下进行苄基溴化反应得到的。4,5用Br2进行苄基溴化存在 一些缺点,每个Br2中都会有一个Br生成副产物HBr,导致Br的利用率很低;而且Br2的毒性和高蒸气压使其在运输和储存方面都存在危险。 6因此,开发了许多试剂和方案来替代Br2,以实现选择性苄基溴化,如H2O2/HBr/NBS,7 BBr3和8 NBS/SiCl4,9还有各种氧化溴化体系,例如NaBrO3/NaHSO3,10 NaBrO3/KBr-/H+,11 KBr/Oxone,12 NaNO2/KBr/HCl13和 HBr/H2O2。14-17其中最值得推荐的氧化溴化体系是HBr/H2O2,因为HBr和H2O2的成本低,Br利用率为100%,H2O是唯一的副产物,这避免了其他氧化剂存在的一些环境问题。18图1. 2,6-二氯苄溴组成的生物活性物质传统上,氧化溴化反应是在间歇式反应器中进行的,14,17但是间歇式反应器存在许多缺点,例如光的辐射距离短,反应效率低,特别是在大规模生产时有爆炸的风险。近年来,随着科学技术的发展,微通道反应器技术在化学转化方面取得了重大进展。19-22与传统的间歇式反应器相比,微通道反应器具有传质和传热效率高、比表面积大、安全性高和操作性好等特点,不仅能精确控制反应条件,获得高选择性的目标产物,而且易于放大。实际上,微通道反应器因其上述优点以及反应物更容易接触到光的优势,在光催化化学领域得到了广泛的应用,包括光氧化催化、23 β-二羰基化合物、24丙烯醛与糖基自由基的共轭加成、25杂环的三氟甲基化、26苯氧化制备苯酚27、光引发的苄基氯化28和脂环化合物氯化。29,30并且微型反应器31-34也被应用于光照下使用Br2或HBr/H2O2进行的苄基溴化反应。另一方面,间歇式反应器中,原位生成的Br2会催化H2O2严重分解,而在微通道反应器中反应物的接触时间较短,所以预计在微通道反应器中用HBr/H2O2进行的氧化溴化反应将会大大减少H2O2的分解。35河北工业大学的张月成教授课题组在荷兰Chemtrix玻璃微通道反应器Kiloflow中,光催化下,以HBr为溴源,H2O2为氧化剂,2,6-二氯甲苯氧化苄基溴化反应制备2,6-二氯苄溴,此过程安全环保,经济高效。并考察了反应温度、反应物料摩尔比、停留时间、光强以及物料浓度等因素的影响,得到了2,6-二氯甲苯制备2,6-二氯苄溴的最佳反应条件。文章发表在ACS omega. 2022, 7, 5, 4624-4629 (https://doi.org/10.1021/acsomega.1c06737)。 一、 【实验部分】2.1实验试剂和仪器所有试剂均为分析级,直接使用,无需进一步纯化。 2,6-二氯甲苯 (DCT)、1,2-二氯乙烷、过氧化氢水溶液 (H2O2, 30.0 wt.%) 和氢溴酸 (HBr, 47.0 wt.%) 由上海泰坦化学试剂合作公司提供。 去离子水是我们实验室自己配制的。在来自 Chemtrix B.V. (Echt, The Netherlands) 的 Kiloflow 型连续流微通道反应器中进行光照射下2,6-二氯甲苯的氧化苄基溴化反应。光源由3个5W的光板和2个36W的灯带组成,发出波长为435-445nm的蓝光。通过将光板和光条放置在距离微通道反应器的玻璃反应板5mm处来照亮反应混合物。在配备有Shim-packVP-ODS C18柱的Shimadzu高效液相色谱仪(HPLC)上分析反应液。 2.2 实验过程在一个典型的实验中,将22.7g(0.141mol)2,6-二氯甲苯和73.0 mL1,2-二氯乙烷依次加入一个锥形瓶中,得到溶液A。在另一个锥形瓶中加入26.0 g 30.0wt.%的H2O2,然后加入75.2 mL去离子水稀释,得到溶液B。然后,在第三个锥形瓶中加入40.0 g 47.0wt.%的HBr,然后加入75.1mL的去离子水稀释,得到溶液C。如图2所示,溶液A、B和C分别由三个10mL的进液泵输送,溶液B和C首先通过一个三通阀混合,并在第二个三通阀中与溶液A混合。混合物最后进入微通道反应器中,在给定的温度和压力下完成反应。反应液从反应器中流出,在冷阱中低温淬灭(0℃)。在整个过程中,反应器的压力由入口处的压力表监测,并由出口处的背压阀控制。 图2. 光照射下的氧化溴化工艺流程反应完成后,反应混合物用高效液相色谱进行分析。首先,用高效液相色谱法(HPLC)建立标准曲线,用于定量分析2,6-二氯甲苯、2,6-二氯苄溴和2,6-二氯苯甲酸。2,6-二氯甲苯的转化率以及2,6-二氯苄溴和2,6-二氯苯甲酸的选择性是用以下公式(1)-(3)计算的: 二、 【实验结果与讨论】3.1 温度对反应的影响对于在微通道反应器中,使用特定波长和强度的光照射2,6-二氯甲苯的氧化苄基溴化反应,反应温度是影响反应的关键因素。因此首先研究了反应温度对反应的影响。经测定,反应主要产物为2,6-二氯苄溴和2,6-二氯苯甲酸,没有观察到芳基取代产物,表明在该反应条件下,微通道反应器对苄基取代反应的选择性很好。如图3所示,随着温度从30℃增加到70 ℃,2,6-二氯甲苯的转化率从15.5%增加到67.8%,2,6-二氯苄溴的选择性从68.7%增加到75.3%。随着反应温度在70℃以上的进一步提高,2,6-二氯甲苯的转化率和2,6-二氯苄溴的选择性都略有增加。同时,在所有情况下,2,6-二氯苯甲酸的选择性几乎保持在10.2%左右。图3.温度对2,6-二氯甲苯氧化溴化反应的影响反应条件:87W蓝光,HBr:H2O2:2,6-二氯甲苯(摩尔比)=1.3:1.3:1;溶液A:15.0wt.%的2,6-二氯甲苯(0.093 mol)溶于73.0 mL1,2-二氯乙烷,1.47 mL/min;溶液B:12.5wt.%HBr水溶液, 1.3 mL/min;溶液C: 5.76 wt.% 的H2O2 水溶液, 1.3 mL/min; 停留时间:5.82 min;反应压力:0.8 MPa。DCT:2,6-二氯甲苯DCBB:2,6-二氯苄溴DCBA:2,6-二氯苯甲酸3.2 HBr和H2O2用量对反应的影响接下来,作者研究了HBr: H2O2:2,6-二氯甲苯的摩尔比对苄基氧化溴化反应的影响。如图4所示,2,6-二氯甲苯的转化率随着HBr:H2O2:2,6-二氯甲苯摩尔比的增加而增加,在1.96:1.96:1时达到95.2%,但2,6-二氯苄溴的选择性随着HBr:H2O2:2,6-二氯甲苯摩尔比的增加而降低;随着其摩尔比的进一步增加,2,6-二氯甲苯的转化率和2,6-二氯苄溴的选择性都略有增加;2,6-二氯苯甲酸的选择性随着HBr: H2O2:2,6-二氯甲苯摩尔比的变化而略有变化,保持在10.4%左右。为了在较低的HBr和H2O2消耗量下获得更高产量的2,6-二氯苄溴,最佳的HBr:H2O2:2,6-二氯甲苯摩尔比被确定为1.5:1.5:1。在这种情况下,2,6-二氯甲苯的转化率为76.1%,2,6-二氯苄溴的选择性为73.8%。以上结果表明,在微通道反应器中用H2O2/HBr进行氧化溴化反应比在间歇式反应器中进行更有优势,因为H2O2的消耗量更低。在间歇式反应器中进行类似的氧化溴化时,H2O2与HBr的最佳摩尔比约为2:1。35图4. (HBr/H2O2): 2,6-二氯甲苯 (摩尔比)对2,6-二氯甲苯氧化苄基溴化反应的影响反应条件: 87 W 蓝光 ;溶液A: 15.0 wt.% 的2,6-二氯甲苯(0.093 mol) 溶于73.0 mL 1,2-二氯乙烷;溶液 B: 12.5 wt.% HBr水溶液; 溶液 C: 5.76 wt.% H2O2 水溶液; 停留时间:5.83 min;反应压力:0.8 MPa; 反应温度:70℃。DCT:2,6-二氯甲苯DCBB:2,6-二氯苄溴DCBA:2,6-二氯苯甲酸3.3停留时间对反应的影响在微通道反应器中,反应物的停留时间是影响反应的另一个关键因素,它也间接反映了光催化反应中光对反应物的照射时间。因此,在 70℃和 HBr:H2O2:2,6-二氯甲苯摩尔比为 1.5:1.5:1 时,研究了停留时间对反应的影响。如图 5 所示,2,6-二氯甲苯的转化率最初随着停留时间的增加而增加,在停留时间为5.88 min 时,转化率达到最大值 76.1%,然后随着停留时间的延长而缓慢下降,直到停留时间达到 9.43 min时,转化率急剧下降。2,6-二氯甲苯转化率在停留时间为9.43min以上时急剧下降,可能是因为在微通道反应器中,水相和有机相在较长停留时间下混合不均匀。 2,6-二氯苄溴的选择性随着停留时间的增加而缓慢增加,在停留时间为5.88 min时达到最大值73.8%,然后随着停留时间的进一步延长而略有下降。与其他情况类似,2,6-二氯苯甲酸的选择性随停留时间略有变化,约为12.7%。因此,停留时间确定为 5.88 min。图5 停留时间对2,6-二氯甲苯氧化苄基溴化反应的影响反应条件:87W蓝光; HBr: H2O2:2,6-二氯甲苯(摩尔比)= 1.5:1.5:1;溶液A: 15.0 wt.% 2,6-二氯甲苯(0.093 mol) 溶于73.0 mL 1,2-二氯乙烷; 溶液B:12.5wt.%的HBr水溶液; 溶液C:5.76wt.%的H2O2水溶液;反应压力:0.8 MPa;反应温度:70 ℃。DCT:2,6-二氯甲苯DCBB:2,6-二氯苄溴DCBA:2,6-二氯苯甲酸3.4光照对反应的影响从表 1 可以看出,2,6-二氯甲苯在黑暗中基本上没有反应,并且在反应液中没有检测到2,6-二氯苄溴(表 1,条目 1)。 当打开光源 ,反应就发生了。 随着光照强度从 72W 增加到 87W,2,6-二氯甲苯的转化率从 66.7% 增加到 76.1%,2,6-二氯苄溴选择性从 76.2% 降低到 73.8%,但2,6-二氯苯甲酸选择性从 9.5% 增加到 12.8%(表 1,条目 2 和 3 )。 随着光照强度的进一步增加,2,6-二氯甲苯转化率缓慢增加,但 2,6-二氯苄溴和2,6-二氯苯甲酸的选择性均下降(表 1,条目 4),这可能是因为在过强的光强度下发生了其他副反应。 因此,确定光强为87W。表1. 光照对2,6-二氯甲苯氧化苄基溴化反应的影响条目条件2,6-二氯甲苯 转化率 /%产物选择性 /%2,6-二氯苄溴2,6-二氯苯甲酸1黑暗---272w 蓝光66.776.29.5387w 蓝光76.173.812.84118w 蓝光78.472.57.6反应条件:HBr:H2O2:2,6-二氯甲苯 (molar ratio) = 1.5:1.5:1; 溶液A: 15.0 wt.% 2,6-二氯甲苯(0.093 mol) 溶于73.0 mL 1,2-二氯乙烷,1.37 mL/min; 溶液B:12.5wt.%的HBr水溶液,1.33mL/min; 溶液C:5.76wt.%的H2O2水溶液,1.33mL/min;停留时间: 5.88 min; 反应压力:0.8 MPa; 反应温度:70℃.3.5物料浓度的影响在HBr: H2O2:2,6-二氯甲苯摩尔比=1.5:1.5:1、停留时间为5.88 min、光照强度为87 W的条件下,考察了物料浓度对反应的影响。 如图6所示,横坐标以2,6-二氯甲苯浓度为基准,随着2,6-二氯甲苯浓度从11.0wt.%增加到 21.0wt.%,2,6-二氯甲苯的转化率从49.8%增加到 98.1%,然后随着物料浓度的进一步增加,转化率缓慢增加。总体而言,2,6-二氯苄溴的选择性随着物料浓度的增加呈现先升高后降低的趋势,在2,6-二氯甲苯浓度为21.0 wt.%时达到最大值93.2%。此外,随着物料浓度的增加,2,6-二氯苯甲酸的选择性呈现逐渐下降的趋势。当2,6-二氯甲苯浓度为21.0wt.%时,2,6-二氯苄溴产率最高,为91.4%。图6 物料浓度对2,6-二氯甲苯氧化苄基溴化反应的影响反应条件: 87W蓝光; HBr: H2O2:2,6-二氯甲苯(摩尔比)= 1.5:1.5:1; 溶液A:2,6-二氯甲苯溶于1, 2-二氯乙烷,1.37 mL/min; 溶液B:HBr水溶液,1.33 mL/min; 溶液C:H2O2水溶液,1.33 mL/min; 停留时间:5.88min;反应压力:0.8 MPa; 反应温度:70℃。DCT:2,6-二氯甲苯DCBB:2,6-二氯苄溴DCBA:2,6-二氯苯甲酸3.6 H2O2对HBr摩尔比的影响优化H2O2/HBr的摩尔比对提高H2O2或Br的利用效率具有重要意义。 在2,6-二氯甲苯、HBr 和 H2O2 浓度分别为21.0wt.%、16.3wt.%和7.7wt.% 以及2,6-二氯甲苯与H2O2为1:1.5 的情况下进行的。 如图7所示,随着H2O2与HBr的摩尔比从1.5:1.5增加到1.5:1.4,2,6-二氯甲苯的转化率和2,6-二氯苄溴的选择性均略有下降,并且随着H2O2与 HBr 的摩尔比的进一步增加呈下降趋势。 然而,在H2O2与HBr的摩尔比为1.5:1.4时,Br的最大利用效率为62.5%。因此,H2O2与HBr的最佳摩尔比为1.5:1.4。图7. H2O2/HBr摩尔比对2,6-二氯甲苯氧化苄基溴化反应的影响反应条件: 87 W 蓝光; 2,6-二氯甲苯:H2O2 (摩尔比) = 1:1.5; 溶液 A: 21.0 wt.%的2,6-二氯甲苯 (0.141 mol)溶于73.0mL 1,2-二氯乙烷; 溶液B: 16.3 wt.% HBr水溶液; 溶液C: 7.1 wt.% H2O2水溶液; 停留时间:5.88 min; 反应压力:0.8 MPa;反应温度:70℃.DCT:2,6-二氯甲苯DCBB:2,6-二氯苄溴DCBA:2,6-二氯苯甲酸基于以上实验结果,得出了光照下微通道反应器中2,6-二氯甲苯氧化苄基溴化生成2,6-二氯苄溴的最佳工艺条件,即HBr: H2O2:2,6-二氯甲苯摩尔比为1.5:1.5:1,2,6-二氯甲苯、HBr和H2O2浓度分别为 21.0 wt.%、16.3 wt.% 和 7.7 wt.%,停留时间 5.88 min,反应压力 0.8 MPa,反应温度 70℃,87 W 蓝光。 在最佳条件下,2,6-二氯甲苯的转化率为98.1%,2,6-二氯苄溴的选择性为93.2%,收率为91.4%。 三、 【结论】以H2O2为氧化剂,HBr为溴源,在光照下的微通道反应器中进行了2,6-二氯甲苯氧化苄基溴化合成2,6-二氯苄溴,此工艺过程安全绿色。在最佳反应条件下,2,6-二氯甲苯的转化率高达98.1%,2,6-二氯苄溴收率为91.4%。微通道反应器可操作性强,反应物在其中更易获得光照,这使反应得以高效进行。 【原文】ACS omega. 2022, 7, 5, 4624-4629 (https://doi.org/10.1021/acsomega.1c06737)
应用实例
2022.04.11

采用荷兰Chemtrix 微通道反应器和微通道固定床反应器实现九步连续流合成奥司他韦
【摘要】纳尔逊·曼德拉大学的C·R·萨甘迪拉P·瓦茨申请的CN 113677658 A发明:生产奥司他韦的流动合成方法提供了由莽草酸生产奥司他韦及其药学上可接受的盐的流动合成方法,特别地但不排他地提供了在九步流动合成中由莽草酸生产磷酸奥司他韦的流动合成方法,该方法与已知方法相比提供了更优的反应时间和产物产率。在九步流动合成中由莽草酸生产磷酸奥司他韦的流动合成方法中涉及酯化、叠氮化、氮丙啶化、开环、酰化及还原等多种类型的反应。除第2,8,9三步反应因成盐生成固体产物需要超声加速移动外,其余六步均可以用荷兰Chemtrix的Labtrix微通道反应器实现连续操作。相对于传统的批次反应,反应时间,转化率,选择性都大幅度提高,而且还为传统的危险反应如叠氮化反应批次操作时不能高温处理,但使用荷兰Chemtrix的Labtrix微通道反应器可以在高温190℃安全操作。磷酸奥司他韦的流动合成案例充分体现了流动合成的可行性,安全,高效,高转化率等优点,给相似的工艺提供了充分的可行性验证,也为后续的生产放大提供了重要参考。【背景技术】流感是一种严重的呼吸系统病毒感染,由于每年的流行和可预测的大流行而导致显著的发病率和死亡率。仅在美国,每年就记录200000人住院和36000人死亡。此外,该病毒每年影响约20%的世界人口,导致约500000人死亡。磷酸奥司他韦是被称为神经氨酸酶抑制剂(NAI)的一类化合物中的一种化合物,用于治疗和预防流感。它对由甲型流感病毒和乙型流感病毒引起的流感有效。现有技术中描述了许多制备磷酸奥司他韦的方法和合成路线。然而,用于生产这些化合物的现有合成方法基本上基于标准的搅拌分批反应器型方法(stirred batch reactor type process),其中使用大量有机溶剂。此外,大多数已知方法要么采用叠氮化物化学,要么采用保护基团化学,这两者特别是在分批方法中都引入了固有的限制。叠氮化物化学因为其危险和高度放热的性质而引起许多安全问题,这在工业规模上变得甚至更加明显。由于这些固有的危险,过程化学家在可以用于最大化反应效率和反应产率的反应参数方面受到限制。另一方面,保护基团化学通常会增加反应时间同时降低总产率,从而增加最终产物成本。最近称为“流动化学”的微型反应器技术(MRT)是一种新兴技术,其使得研究和开发人员能够利用连续流动快速筛选反应,从而确定适合在生产水平上使用的反应条件。此外,除了使用传统的反应方法以外,与使用小反应器体积相关的固有安全性使得使用者能够采用以前认为在生产环境中使用过于危险的反应条件;如极端的反应条件或使用/生成“危险”化合物。因此,通过使用该技术增加了化学家可用的反应类型。此外,在磷酸奥司他韦的情况下,连续流动合成可能提供一种足够有效的技术,使得能够在大流行的情况下特别是在发展中国家实现快速本地制造。纳尔逊·曼德拉大学的C·R·萨甘迪拉P·瓦茨申请的CN 113677658 A发明:生产奥司他韦力图通过提供用于生产奥司他韦的新型流动化学方法,来解决现有技术的一些缺点。【流动反应器】2.1 Labtrix微通道反应器(品牌:荷兰Chemtrix)Labtrix微通道反应器(品牌:荷兰Chemtrix)是一种手动操作的“即插即用”连续流动反应器系统,用于在微型反应器内进行快速反应筛选和方法优化。该系统具有模块化设置,其便于更换组件以增加化学兼容性、进料管线数量或反应器类型或体积。它可以用于在-20℃至195℃的温度范围和20bar的最大操作压力下使用非常少的试剂进行反应。该系统主要由Labtrix启动单元、热控制器、注射泵、注射器、管道和配件组成。启动单元容纳微型反应器。它可以加热或冷却至介于-20℃至+195℃之间的温度,由热控制器控制。装有玻璃气密性Leur锁注射器的注射器泵将试剂计量至微型反应器中。该系统有十二种不同的可互换玻璃微型反应器类型,其体积和设计各不相同。这些玻璃反应器根据其设计和混合模式分为三个不同的类别。T?混合反应器、SOR-混合反应器和催化剂反应器是所述三个不同的类别。微通道反应器(品牌:荷兰Chemtrix 型号:Labtrix Start)2.2 微通道固定床反应器系统[0086] 该系统由带有增强PEEK可调端接头的10mm i .d .×100mm Omnifit玻璃柱组成。使用PTFE管(0 .8mm ID)将柱反应器连接至HPLC泵,并从反应器连接至收集容器。使用流速范围为0 .00-10 .00ml/min的蠕动HPLC泵系列III(10ml泵头),将液体试剂计量通过装有10bar背压调节器的固定床反应器。(一正科技微通道固定床反应器)一正科技微通道固定床反应器2.3 超声处理PTFE盘管反应器(Coil Reactor)系统超声处理PTFE盘管反应器(Coil Reactor)系统该系统由装有两个装有试剂的10ml SGE玻璃注射器的Chemyx注射泵组成。将两股试剂流泵入T-混合(Omnifit labware,孔径:8 .0mm ID,0 .5-4mm OD) ,其连接至0 .8mlPTFE盘管反应器(0 .8mm ID,1 .6ml管长),下游有产物收集瓶。将T-混合器和PTFE盘管反应器放入温度控制的超声水浴中。EINS SCI专业超声波浴(40kHz)用于超声处理。【连续流反应过程】图1 用于生产磷酸奥司他韦的连续流动合成方法的优化合成路线示例3.1 反应1:莽草酸8的连续流动酯化莽草酸8的酯化是合成(-)-磷酸奥司他韦的第一步(方案1)。研究了各种酯化条件以优化Chemtrix Labtrix微通道反应器和填充床柱流动系统中的酯化反应。分别在Chemitrix Labtrix微反应器系统和固定床反应器中进行所有溶液相和固相酯化反应。Chemtrix Labtrix微反应器系统用于进行所有溶液相酯化的研究。该系统装有19 .5μl玻璃反应器,用于在催化剂存在下优化莽草酸酯化。亚硫酰氯、草酰氯、亚硫酰氯/DMF、草酰氯/DMF、苯磺酸(BSA)和对甲苯磺酸(PTSA)是用于莽草酸酯化的研究的各种催化剂。两个注射泵用于将试剂从两个10ml SGE Luer锁气密玻璃注射器泵入装有10bar背压调节器的热控制微型反应器系统中。莽草酸(0 .1M)和催化剂均溶解于乙醇中,并分别泵入流动系统。使用HPLC方法A收集和分析样品。表1 Chemitrix Labtrix系统操作反应1的反应效果酯化催化剂反应当量比反应温度停留时间转化率SOCl21:1140℃8min93%(COCl)21:21608min99%BSA10:119020min94%PTSA10:119040min96%备注:1.反应当量比为草莽酸:催化剂 2.批次反应时间3h为了避免生成大量的酸废物,研究了在连续流动系统中使用固体酸催化剂Amberlyst 15与乙醇进行莽草酸酯化。装有固体催化剂的微通道固定床反应器用于所有固相酯化研究(方案2)。向10mm ID x 100mm 玻璃柱内填充Amberlyst 15或Amberlyst(3cm床高,2 .4反应器体积)。对柱反应器进行热控制,并使用10bar背压调节器对系统加压。使用蠕动HPLC泵将莽草酸的乙醇溶液(0 .1M)泵入加热的填充床中。使用HPLC方法A收集和分析样品。在干燥的Amberlyst 15作为莽草酸酯化催化剂的存在下,莽草酸转化率随着温度和停留时间的增加而增加。在实验设置中,发现最佳条件是140℃和8min停留时间,以得到92%的莽草酸转化率。这与使用SOCl2进行莽草酸酯化的最佳条件(93%,140℃和8min停留时间)相比更有利。此外,从健康、环境和安全的角度来看,Amberlyst 15程序比危险的SOCl2程序更受欢迎。作为额外的优势,可以在反应结束时去除Amberlyst 15,并再生以供进一步使用。3.2 反应2:莽草酸乙酯39的连续流动甲磺酸化由于在反应过程中MsCl和TEA之间形成的铵盐沉淀,在玻璃微通道反应器(荷兰Chemtrix,型号:Labtrix)的 SOR3227芯片(19 .5μL)反应器,(300μm通道宽度,120μm通道深度)中,0 .8mm更大通道直径的玻璃反应器,简单PTFE盘管反应器(1mm ID)进行实验,即使在非常低的浓度下,反应器都会出现堵塞的问题。但在超声处理下的0 .8ml PTFE盘管反应器(0 .8mm ID,1 .6m管长) (方案4)不会发生堵塞问题。超声处理看来有助于铵盐沉淀的移动,从而避免了反应器堵塞。因此,这一发展使得我们能够研究不同的反应参数并最终优化反应。然而,可以设想当反应扩大到工业规模时可能不需要超声处理。。使用超声处理下的0 .8ml PTFE盘管反应器(0 .8mm ID,1 .6m管长)优化莽草酸乙酯39的甲磺酸化,以得到三甲磺酸酯40。将莽草酸乙酯39(0 .2M)在乙酸乙酯中与甲磺酰氯(0 .9M,4 .5当量)预混合,以制备第一溶液。莽草酸乙酯39不易溶于乙酸乙酯。因此,首先将其溶解于热乙酸乙酯中,再冷却,然后与甲磺酰氯预混合。通过将有机碱溶解于乙酸乙酯中来制备第二溶液。筛选了以下的碱:三乙胺(TEA)、咪唑、1 ,8-二氮杂双环[5 .4 .0]十一碳-7-烯(DBU)、1 ,4-二氮杂双环[2 .2 .2]辛烷(DABCO)和三己胺(THA)。首先通过PTFE注射式过滤器(0 .45μl孔径)过滤收集的样品,以去除反应过程中形成的铵盐,然后使用HPLC方法B进行分析。由于停留时间和反应温度的增加导致不显著的莽草酸乙酯转化率,因此研究了增加碱(TEA)浓度的影响。这些实验在室温、12s停留时间下使用莽草酸乙酯(0 .2M,1当量)、MsCl(1 .5当量),同时改变TEA浓度。这些实验的结果示于图2。从图2可以看出,莽草酸乙酯转化率随着碱(TEA)浓度的增加而增加。发现最佳条件是在室温和12s停留时间下使用莽草酸乙酯(0 .2M)、MsCl(0 .9M,有效1 .5当量)、TEA(3M,15当量),以100%的转化率得到所需的甲磺酸酯。观察结果表明,反应甚至可以在比12s短得多的停留时间下进行。然而,由于可用的注射泵的限制,难以全面研究较低的停留时间。据报道,在低温优选在0℃,在三乙胺(TEA)作为碱存在下,使用MsCl以分批式反应进行莽草酸乙酯39的甲磺酸酯化,进行约2至4小时。图2 反应2转化率与TEA浓度关系图图3 反应2中使用不同催化剂的转化效率从图3可以看出,在研究的碱中TEA转化率最高,而DABCO表现最差。除了DABCO,所有研究的碱都给出了与TEA相当的结果。仍然存在铵盐沉淀问题。然而,观察到使用DBU和咪唑时沉淀较轻。有趣的是,使用THA得到了澄清溶液。不存在沉淀可以归因于(THA)疏水性的增加,由于与TEA相比链长增加,使得所形成的铵盐可溶于反应溶剂乙酸乙酯中。3.3 反应3:(3R ,4S ,5R)-3 ,4 ,5-三-O-甲磺酰莽草酸乙酯40的连续流动叠氮化通过使用不同的叠氮化试剂和条件,将烯丙基C-3位置的OMs基团立体选择性和区域选择性亲核取代为叠氮基(方案5)。在各种叠氮化剂的存在下,采用玻璃微通道反应器(荷兰Chemtrix,型号:Labtrix)的 SOR3227芯片(19 .5μL)反应器,以优化甲磺酰莽草酸酯40的烯丙基C-3位置的OMs基团的叠氮化(方案6)。叠氮化钠(NaN3)、叠氮磷酸二苯酯(DPPA)、叠氮化三甲基甲硅烷(TMSA)和叠氮化四丁基铵(TBAA)是该系统中研究的各种叠氮化剂。必要时使用HCl水溶液(0 .11M,1 .1当量)在流动反应器内淬灭反应。使用HPLC方法A收集和分析样品。1 .1当量的NaN3、50℃和12s停留时间,得到向所需的叠氮化物41的完全转化。尽管如先前报道的,高温、长反应时间和碱度不利于对分批的所需的叠氮化物41的选择性,但从我们的实验中可以明显看出,使用微型反应器显著提高选择性,更大幅减少反应时间。与所有公开的文献程序相反,使用我们的程序不生产副产物。3.4 反应4:(3S ,4R ,5R)-3-叠氮基-4 ,5-双(甲磺酰氧基)环己-1-烯羧酸乙酯41的连续流动氮丙啶化在Chemtrix Labtrix微通道反应器(方案9)中进行叠氮化物41的连续流动氮丙啶化。采用玻璃微通道反应器(荷兰Chemtrix,型号:Labtrix)的 SOR3227芯片(19 .5μL),以优化使用亚磷酸三烷基酯的叠氮莽草酸酯41的氮丙啶化反应。亚磷酸三乙酯和磷酸三甲酯是研究的两种亚磷酸烷基酯。分别使用两个注射泵,将叠氮莽草酸酯的无水乙腈溶液(0 .1M)和亚磷酸三烷基酯的无水乙腈溶液(0 .11M ,1 .1当量)从两个10ml SGE Luer锁气密玻璃注射器泵入装有10bar背压调节器的热控微型反应器系统中(方案9)。使用HPLC方法A收集和分析样品。用(EtO)3P或(MeO)3P (0 .11M,1 .1当量)处理叠氮化物41 (0 .1M)的乙腈溶液,以得到氮丙啶42,结果表明,氮丙啶的形成随着温度和停留时间的增加而增加,在大约190℃和3s停留时间下,使用(EtO)3P和(MeO)3P分别形成了93%和98%的氮丙啶42。重要的是,此发明中的系统和方法允许高温叠氮化物化学,微型反应器允许在非常高的温度下安全地探询潜在的爆炸性叠氮化物化学,与先前报道的5小时分批反应相比反应非常迅速。3.5 反应5:(3R ,4S ,5R)-4-(二乙氧基磷酰氨基)-5-甲磺酰氧基-3-(戊-3-基氧基)环己-1?烯羧酸乙酯43的连续流动合成在连续流动系统中,氮丙啶42与3-戊醇和路易斯催化剂三氟化硼乙醚络合物在烯丙基位置进行区域和立体选择性开环(方案10)。采用玻璃微通道反应器(荷兰Chemtrix,型号:Labtrix)的 SOR3227芯片(19 .5μL),以优化用3-戊醇和三氟化硼乙醚络合物的氮丙啶42开环(方案11)。分别使用两个注射泵,将乙腈/3-戊醇(50:50)中的氮丙啶42(0 .1M)和三氟化硼乙醚络合物(0 .15M ,1 .5当量)的乙腈/3-戊醇(50:50)溶液从两个10ml SGE Luer锁气密玻璃注射器泵入装有10bar背压调节器的热控微型反应器系统中。使用HPLC方法A收集和分析样品。由氮丙啶42向3-戊醚43的转化率随着停留时间和温度的增加而增加。温度升高导致转化率显著提高。在12s停留时间下,在25℃和100℃分别实现3-戊醚43产率66%和100%。发现优选的条件是约100℃和12s停留时间,以得到向3-戊醚43的完全转化。3.6 反应6:(3R ,4S ,5R)-4-乙酰氨基-5-甲磺酰氧基-3-(戊-3-基氧基)环己-1-烯羧酸乙酯44的连续流动合成通过用硫酸裂解N-P键,然后在弱碱性条件下乙酰化,实现了3-戊醚43的乙酰化(方案12)。通过用乙腈中的H2SO4(0 .8M,8当量)处理乙腈中的3-戊醚43(0 .1M),在第一热控反应器中原位形成中间体43a。在第二热控反应器中用NaOH(1 .62M,16 .2当量),然后用乙酸酐(1 .6当量)处理原位形成的中间体43a,以得到乙酰胺44。这种用于多级连续流动系统的系统装有10bar的背压调节器。使用HPLC方法A收集和分析样品。3-戊醚43向化合物43a的转化率随着温度和停留时间的增加而增加,连续流动N-P键断裂的优选条件是约170℃和3s停留时间,使用H2SO4(8当量)以得到化合物43a的完全转化。通过随后在弱碱性条件下用Ac2O处理原位形成的化合物43a,以在连续流动系统中得到乙酰胺44,来完成3-戊醚43的乙酰化(方案13),在室温用NaOH水溶液(0 .8M,16 .2当量)、再用Ac2O(0 .8M,1 .6当量)处理化合物43a,30s总停留时间得到93%的乙酰胺44,超过30s停留时间转化率没有提高。3.7 反应7:(3R ,4S ,5S)-5-叠氮基-4-乙酰氨基-3-(1-乙基-丙氧基)-环己-1-烯羧酸乙酯32的连续流动合成用合适的叠氮化剂处理乙酰胺44,以得到叠氮化物32。乙酰胺44上的C-5OMs基团被N3基团亲核取代(方案14)。使用装有19 .5μl玻璃反应器的Chemtrix的Labtrix启动连续流动系统,以优化乙酰胺44的C-5叠氮化(方案15)。分别使用两个注射泵,将乙腈中的乙酰胺44(0 .1M)和适当溶剂中的叠氮化剂(0 .3M,3当量)从两个10ml SGE Luer锁气密玻璃注射器泵入装有10bar背压调节器的热控微型反应器系统中。研究了NaN3、TBAA、DPPA和TMSA的使用。使用HPLC方法A收集和分析样品。叠氮化物32的形成是温度和停留时间的函数。乙酰胺44向叠氮化物32的转化率随温度升高而增加。在45s停留时间下,向叠氮化物32的转化率在80℃和190℃分别为55%和100%。发现优选的条件是约190℃、45s停留时间,以得到叠氮化物32的完全转化。在19 .5μl玻璃微型反应器中,为NaN3开发的优选条件(3当量、190℃和45s)用于研究使用DPPA、TMSA和TBAA作为乙酰胺44的叠氮化剂。在这些实验中,乙酰胺44在DPPA、TMSA和TBAA作用下以不同的转化率成功转化为叠氮化物32。看来,应用离子键合叠氮化物(NaN3和TBAA)得到相似的转化率(分别为100%和93%),而共价键合叠氮化物(DPPA和TMSA)导致相对较低的转化率(分别为84%和81%)。3.8 反应8:奥司他韦33的连续流动合成使用超声处理下的0 .8ml PTFE盘管反应器(0 .8mm ID,1 .6m管长) (方案17)来优化使用NaBH4和CoCl2的叠氮化物32的还原,以得到奥司他韦。将叠氮化物32(0 .15M)与乙醇中的CoCl2(0 .1当量)和水中的NaBH4(0 .30M,2当量)的混合物(pH=8)泵送通过连续流动系统,以得到奥司他韦33。首先通过PTFE注射式过滤器(0 .45μl孔径)过滤收集的样品,以去除反应中形成的硼化钴沉淀,然后使用HPLC方法A进行分析。向奥司他韦33的转化率随着停留时间的增加而增加。出人意料地发现,在仅1s和5s停留时间下,向奥司他韦33的转化率分别为81%和96%。发现优选的条件是大约室温和大约5s停留时间,以得到奥司他韦33(96%)。3.9 反应9:磷酸奥司他韦3的连续流动合成在连续流动系统中用H2PO4处理奥司他韦33,以得到磷酸奥司他韦3(方案18)。在连续流动系统中,使用超声处理下的0 .8ml PTFE盘管反应器(0 .8mm ID,1 .6m管长)来优化用H2PO4处理奥司他韦33,得到磷酸奥司他韦3。将乙醇中的奥司他韦33(0 .1M)和乙醇中的H2PO4 (0 .12M,1 .2当量)泵送通过热控连续流动系统,以得到磷酸奥司他韦。使用HPLC方法A收集和分析样品。在50℃、在超声处理下的0 .8ml PTFE盘管反应器(0 .8mm ID,1 .6m管长)中,用乙醇中的H2PO4 (0 .12M,1 .2当量)在不同停留时间下处理乙醇中的奥司他韦33(0 .1M)以进行优化。向磷酸奥司他韦3的转化率随着停留时间的增加而增加。优选的条件是约50℃和60s停留时间,以得到磷酸奥司他韦3(98%,HPLC),这是对任何先前报道的反应的显著改进。【结论】在九步流动合成中由莽草酸生产磷酸奥司他韦的流动合成方法中涉及酯化、叠氮化、氮丙啶化、开环、酰化及还原等多种类型的反应,除第2,8,9三步反应因成盐生成固体产物需要超声加速移动外,其余六步均可以用Chemtrix的labtrix微通道反应器实现连续操作。另外,相对于传统的批次反应,反应时间,转化率,选择性都大幅度提高。而且还为传统的危险反应如叠氮化反应批次操作时不能高温处理,但使用Chemtrix的labtrix微通道反应器可以在高温190℃安全操作,详见表2。磷酸奥司他韦的流动合成案例充分体现了流动合成的可行性,安全,高效,高转化率等优点,给相似的工艺提供了充分的可行性验证,也为后续的生产放大提供了重要参考。反应步骤反应类型流动反应效果釜式反应现状反应温度反应时间转化率反应1酯化140℃8min93%反应时间长,3h反应2酯化室温12s100%0℃,反应时间长2-4h反应反应3叠氮化50℃12s100%危险,不能高温操作,反应时间长,选择性低反应4氮丙啶化190℃3s93%危险,不能高温操作,反应时间5h反应5开环100℃12s100%转化率低反应6N-P键断裂/酰化170℃/室温6s/30s100%/93%转化率低反应7叠氮化190℃45s100%危险,不能高温操作,反应8还原室温5s96%黑色沉淀,转化率低反应9磷酸化50℃60s98%反应时间长,转化率低【专利原文】【1】生产奥司他韦的流动合成方法,C·R·萨甘迪拉P·瓦茨,纳尔逊·曼德拉大学,CN 113677658 A【一正科技简介】作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰ChemtrixB.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件: /include/upload/kind/file/20220107/20220107190354_0546.pdf
应用实例
2022.03.07
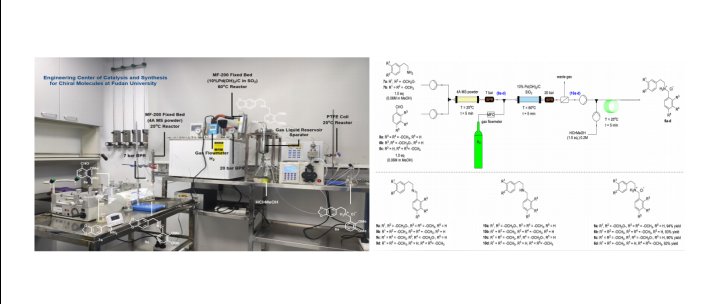
【复旦连续加氢案例】采用一正科技MF-200微通道固定床反应器实现连续加氢案例—— 应用对映选择性催化氢化实现四氢小檗碱类生物碱的全合成研究
【背景介绍】 (-)-四氢小檗碱(1),(-)-四氢巴马汀碱(2),(-)-四氢表小檗碱(3)和(-)-四氢伪巴马汀碱(4)均是从延胡索和千金藤属植物中分离出来的1,是天然四氢原小檗碱类生物碱家族成员,如图1所示。从结构上看,四氢原小檗碱生物碱包含一个5,8,13,14-四氢-6H-二苯并[a,g]喹啉嗪四环体系,在C-14位置具有一个立体中心,在A环的C-2、C-3位置以及D环的C-9、C-10或C-10、C-11位置均具有含氧官能团。 这类生物碱具有多种药理活性,如:抗菌2、抗炎3、抗精神病4和镇痛活性5,而且其中一些化合物可作为a1A肾上腺素受体、多巴胺D2/D1受体及5-HT1A受体的高效、高选择性抑制剂,已引起人们广泛关注。因此,这类生物碱已被视为开发a1A肾上腺素受体、多巴胺D2/D1受体和5-HT1A受体新拮抗剂的先导化合物6。因此,专家们建立了该类生物碱衍生物及结构类似物的活性筛选程序,并全力投入临床试验4,6a,6b,7。 [图1. 四氢原小檗碱类生物碱的代表性实例] 由于该生物碱家族1-4独特的分子结构显示出具有良好的生物活性但自然资源含量低,因此作者对四氢原小檗碱类生物碱的不对称全合成产生了浓厚的兴趣。到目前为止,专家们已经开发出了多种方法来解决四氢原小檗碱1-4的不对称全合成(方案1)8。这些工作包括: (i)Schore通过手性Meyers'甲脒辅助基诱导取代和硅酰定向Pictet-Spengler环化实现1-3的合成(总收率高达51%,55-60% ee)8a。 (ii)Enders以3,4-二甲氧基苯甲醛为原料采用具有RAMP导向的1,2-加成/环化七步合成化合物2(总收率17%,89%ee)8b。 (iii)由Mastranzo课题组开发的两条高度对映选择性路线,包括亚砜基定向加成/环化和亚砜基诱导的Pictet-Spengler反应,分别通过使用芳基取代亚砜和芳基亚砜酰亚胺作为底物获得1-2(总收率高达30%)和4(总收率52%)8c,8d。 (iv)Davis研究的不对称全合成4(总收率18%)依赖于手性亚砜基辅助基诱导的加成/环化反应8e。 (v)Doye利用环亚胺的Noyori不对称转移氢化反应(ATH)合成了4(总收率55%,ee 93%)8f。 (vi)Tong通过不对称Redox-A3反应(总收率高达4%,ee值为88-97%)和基于对映体Noyori催化不对称转移氢化(ATH)(总收率高达12%,ee为77-99%)实现了1-4的两种催化合成方法8g。 虽然这些研究取得了很大进展,但仍有提高合成效率的空间,例如控制获得更高的对映选择性和寻找更有效的方法来建立四个核心环。在本文中,作者报道了一条不对称全合成通用策略来合成四氢原小檗碱类生物碱化合物,包括(-)-四氢小檗碱(1),(-)-四氢巴马汀碱(2),(-)-四氢表小檗碱(3)和(-)-四氢伪巴马汀碱(4)。 [方案1. 以前的不对称合成1-4路线] 复旦大学化学系陈芬儿院士课题组研究发表了一条不对称全合成四氢原小檗碱类生物碱((-)-四氢小檗碱,(-)-四氢巴马汀碱,(-)-四氢表小檗碱和(-)-四氢伪巴马汀碱)的简明路线。文章发表在J. Org. Chem. 2021, 86, 8143?8153(https://doi.org/10.1021/acs.joc.1c00602)。 文章实现了以上四种四氢原小檗碱类化合物的全合成,该路线由市售二取代苯乙胺和二取代苯甲醛为起始原料,经以下步骤实现: 第一步:通过全连续流技术实现了仲胺盐酸盐的高效和可持续合成; 第二步:本文开发了Pictet-Spengler反应/Friedel-Crafts羟基烷基化/脱水级联反应,用于构建二氢原小檗碱的四元环核心结构(ABCD环); 最后一步:进行铱金属催化的对映选择性氢化反应,用于在四氢原小檗碱类生物碱的C-14位置引入所需的立体化学结构。 本项研究大大加快了整个四氢原小檗碱类生物碱家族以及其结构多样性的非天然类似物的不对称合成。 【实验分析】 四氢原小檗碱生物碱1-4的逆合成分析如方案2所示。由于过渡金属催化烯胺的对映选择性氢化这一新兴领域,可作为不对称合成中构建手性环叔胺的有力工具9。在这些四氢原小檗碱天然产物的C环上的C-14位置上具有一个立体手性中心,1-4可通过最后铱金属催化的环烯胺对映选择性氢化反应在二氢小檗碱5中进行建立。二氢原小檗碱5四环母核的环化可尝试通过仲胺盐酸盐6和乙二醛10进行Pictet-Spengler/Friedel-Crafts羟基烷基化/脱水级联反应实现10,该问题有待本文解决。所需的盐酸胺6可在连续流动条件下通过三个步骤由简单的市售二取代苯乙基胺7和二取代苯甲醛8进行获得11。 方案2. 1-4的逆合成分析 本文的合成从相应的市售二取代苯乙胺7a-7b和二取代苯甲醛8a-8c在连续流动中完全连续流动合成仲胺盐酸盐化合物6a-6d开始(方案3)。如方案3所示,通过T型混合器将7a-7b的甲醇溶液与8a-8c混合后泵入MF-200微通道固定床反应器(深圳市一正科技有限公司),该反应器填充有4A MS粉末(2 ml内部体积),在室温和7 bar背压的条件下,在5分钟的停留时间后,分别得到了亚胺9a-9d。 将反应器流出物与H2一起进入另一个含有10%Pd(OH)2/C的固定床反应器进行催化加氢反应,该反应器由SiO2(5 ml内体积)在60℃和20 bar背压条件下进行分散,停留时间为5 min,得到相应的仲胺10a-10b。将反应液与0.2 M盐酸甲醇溶液混合,并在室温下将其引入PTFE反应器盘管(5 ml,内径0.8 mm),停留时间为5 min,以高达90-94%的总收率获得所需的仲胺盐酸盐化合物6a-6d。 [方案3. 仲胺盐酸盐的全连续流动合成6a?6d] 在拥有足够量的构建模块6a-6d的情况下,本文尝试研究Pictet-Spengler反应/Friedel-Crafts羟基烷基化/脱水级联反应,以获得二氢原小檗碱5a-5b。目前还没有研究报道通过这种新的级联反应使用仲胺衍生物和乙二醛合成二氢原小檗碱的实例(表1)。首先,本文在60℃和无水AlCl3存在的条件下,将6a和过量乙二醛混合在二氯甲烷(DCM)中反应。不幸的是,该级联反应只产生了微量的产物5a(条目1)。再尝试其他酸作为添加剂,如TfOH和98%的HCOOH,通过1HNMR分析,也仅产生了微量的产物5a,并且在后处理时未观察到任何产物的出现(条目2-3)。 如果在相同条件下使用TFA代替AlCl3、TfOH和98%HCOOH,反应可生成少量5a(产率10%,条目4)。当单独使用过量98%HCOOH作为溶剂时,对5a的收率没有进一步提高(条目5)。当反应温度增加到80℃时,5a的产率在表1条目6所示的条件下提高到了30%。另外,6a与2.0当量的硼酸在80℃温度下的98%HCOOH中反应12小时时,最终以50%的产率生成5a(条目7)。当进一步添加2.0当量的硼酸和3.5当量的NaCl作为添加剂时,98%HCOOH作为溶剂,反应温度80oC,反应时间12h,该条件下可以71%分离产率收获所需产物5a(条目8),该条件为反应的最适条件。在最佳反应条件下,该级联反应还应用于其他三种仲胺盐酸盐取代物6b-6d,从而相应的得到二氢原小檗碱衍生物5b-5d(条目9-11)。 表1. 不同条件下二氢原小檗碱5a?5d的合成 如前文所述,这项工作最具挑战性的部分是不对称合成四氢原小檗碱类生物碱分子,如何在C-14位置构建立体中心。5a-5d通过铱金属催化的对映选择性氢化就是一种建立这种C-14手性中心的有效可行的策略,进而完成了四氢小檗碱,四氢巴马汀碱,四氢表小檗碱和四氢伪巴马汀碱(1-4)的不对称全合成。首先,本文基于最近报道的通过铱金属催化不对称亚胺氢化/内酰胺化级联反应全合成(?)-20-反式-长春胺和(?)-20-反式-象牙酮宁的反应条件,对二氢小檗碱5a进行铱金属催化对映选择性氢化反应条件进行筛选,以评估手性配体L1-L7(表2)12,13。 在室温80 atm H2的条件下,使用碘化钾作为添加剂,在甲苯/乙酸(v:v=9:1)溶剂中,由0.5 mol%的[Ir(COD)Cl]2和2.0 mol%(R)-MONOPHOS配体L1原位生成的1.0 mol%Ir/配体L1络合物催化5a加氢,反应在3天内完成,但产物为(R)-5a具有与天然产物四氢小檗碱(1)相反的构型,具有较高的产率和中等的对映选择性(91%,-46%ee,条目1)。而在配体L2-L6(条目2-6)的条件下,几乎没有观察到对映选择性。所以接下来的研究集中在二茂铁基配体L7上,实验证明该类配体是催化加氢的最优配体,获得产品的产率为94%,ee为82%(条目7)。当用I2、TBAI或LiI替代KI作为添加剂时,可以显著提高四氢小檗碱(1)(条目8-10)的对映选择性水平。其中KBr的效果最好(92%,96%ee,条目11)。 为了进一步探讨溶剂在加氢反应中的促进作用,作者筛选了几种溶剂体系。然而,当使用MeOH/AcOH(9:1)时,获得了中等的对映选择性(48%ee)(条目12)。三氟乙醇/AcOH(9:1)混合溶剂系统尽管产率较高,但在对映选择性方面效果不佳。令人高兴的是,EtOAc/AcOH(9:1)和DMF/AcOH(9:1)在产率和对映体比率方面表现出非常好的性能(分别为87%、94%ee和88%、94%ee,条目14-15)。但DCM/AcOH的比例为9:1时提供了最佳的结果,即得到最佳的产率和最高的对映选择性(93%,99%ee,条目16)。 此外,使用0.1%的铱金属催化剂进行了5a的克级反应,并以92%的产率和98%的ee(条目17)获得了所需产物四氢小檗碱(1),表明铱金属催化氢化是可靠和实用的。接下来,本文优化的氢化条件扩展应用到了合成其他二氢原小檗碱5b-5d,最终以优异的产率和对映选择性完成了相应的四氢原小檗碱生物碱四氢巴马汀碱(2),四氢表小檗碱(3)和四氢伪巴马汀碱(4)的全合成(分别为95%,97%ee;90%,92%ee;91%,91%ee。条目18-20)。合成(–)-1-(–)-4的1HNMR、13CNMR、FT-IR和比旋度分别与原始相应的分离天然产物的记录相匹配(更多详情见附录)14。 表2. 通过Ir催化的对映选择性氢化5a?5d完成1-4的全合成 【结论】 1、从二取代苯乙胺7和二取代苯甲醛8开始,四氢原小檗碱生物碱(1-4)的全合成分三步完成。该合成的关键步骤是通过Pictet-Spengler反应/Friedel-Crafts羟基烷基化/脱水的级联环化和铱金属催化的不对称氢化反应。 2、 这项工作证明了Pictet-Spengler反应/Friedel-Crafts羟基烷基化/脱水级联环化反应可高效构建二氢原小檗碱生物碱的四元母核,以及铱金属催化烯胺的对映选择性氢化可简洁地引入C-14位的绝对立体构型。 3、本文的合成策略可以适用于其他相关的四氢原小檗碱类生物碱和更多相关的非天然类似物的不对称结构的构建。 4、 作者所在的复旦大学陈芬儿院士课题组目前正在进一步研究该统一策略对其他生物活性的生物碱家族及其衍生物的适用性,研究工作将在不久后发表。 【原文】 The Journal of Organic Chemistry 2021, 86, 8143?8153 https://pubs.acs.org/doi/10.1021/acs.joc.1c00602 【一正科技简介】 作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。 公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。 公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。 公司网址:www.e-zheng.com 联系电话:0755-83549661 产品热线:400-0755-403 应用原文及附件请下载下方PDF文件: /include/upload/kind/file/20211228/20211228144602_0594.pdf
应用实例
2022.01.06

【KU Leuven案例】Chemtrix微反应器中研究光催化反应——气液流动中的光子输运和流体动力学(微反应器中泰勒流动的表征)
【实验背景】气液光反应是流动反应器中光化学反应的重要组成部分。大多数气液光反应主要是在微反应器中研究的,因为微反应器的穿透深度较小,并且可以促进分段流动(通常称为塞流或泰勒流)。这种流动模式包括液塞的交替(an alternation of liquid slugs),润湿通道壁的相(连续相)和气泡(分散相),如图1所示。在气泡和反应器通道壁之间形成了一个薄的液膜。与气液间歇式反应器相比,泰勒流动具有混合和界面传质增强、安全性提高的特点。 在具有活性气相的气液光化学反应中,光敏化的单线态氧的加成近年来受到越来越多的关注,并被应用于合成作为香料的商业化学产品(例如,由β-柠檬烯醇合成芳香玫瑰氧化物)和药物(例如,由二氢青蒿酸合成抗疟青蒿素)。此外,含有非反应性气体的气液泰勒流光反应器,例如氮气与使用单一液相相比被用来实现更大的转换效果。通过延长光化学反应的停留时间(如增加反应器通道长度或降低液流量)和增加光源的强度,可以提高光化学反应的转化率。虽然这些策略受光反应器和泵特性的限制,但加入一个惰性相可以让困难的光化学过程的更易获得。图1说明了产生这种改进的因素: (i) 存在接收高光子通量的薄液膜, (ii) 界面处的光散射, (iii) 在液塞中以两相流混合。图1 在非反应气体的气液流动中,影响反应重要因素的示意图Nakano等人在存在氮气气泡的情况下研究了羰基化合物与烯烃的光环加成Paterni-Büchi型光反应。通过增加气体分数,气泡长度比液塞的长度相对增加。与单液相相比,当气泡与液塞的长度之比在1:1~1:4之间变化时,其转化率在有所提高。产品产量也出现了类似的增长。此外,他们还观察到,当甲苯被三甲基苯取代时,两相的收率从80%左右下降到45%左右,这是一种更粘稠的溶剂。他们认为这种现象是由于液体混合的减少和粘度的增加。以前,他们在甲苯-水泰勒流中对相同类型的反应进行了研究。在这种情况下,甲苯是润湿通道的相,水是无反应的分散相。讨论了水含量对观测结果的影响。与单液相光化学反应相比,当水份为0.2时,两相流动的转化率提高了6.5%,当水份为0.8时,两相流动的转化率提高了77%。Nakano等人进行了另外一项研究,详细分析了光源、浓度和溶剂的影响。此外,Telmesani等人用水作惰性相,研究了在乙酸乙酯-水泰勒流动中肉桂酸的光环加成。在两相流中,当水份为0.9时,两相流的转化率为43%,而当水份不存在时,两相流的转化率为6%。水的加入使反应时间减少到2小时以下,而单相反应的典型反应时间为8小时左右。与以前的研究类似,他们观察到随着水份分数的增加,由于含水的菱形柱,使混合得到改善,转化率增加。此外,当水被全氟化溶剂代替时,有机段塞就变成了分散相。在这种新的流动模式中,有机相为连续相时,转化率下降到19%,而有机相为连续相时为43%。本实验证明,在液膜中存在试剂是至关重要的。上述研究表明,采用惰性不相容相的方法是进行光反应是可行的,并给出了影响转化的参数。为了促进这种方法的进一步实现,需要有更多的知识来预测最佳的反应条件。由溶剂的粘度、表面张力、反应器壁的润湿性、气液流量等因素决定了气泡和液塞长度、膜厚、液塞混合等流动规律的特点。虽然以前在质量输运研究的背景下对流动特性进行了分析,但它们对光子输运的影响尚未系统地研究。光反应器通道中的光吸收可以用化学光度法进行实验定量。在微反应器中实现的大多数光度测量都是在单相流中进行的。虽然用铁氧酸盐化学光度计对环流进行了研究,但以前从未讨论过气液泰勒流中光的吸收。此外,由于气液界面处的反射率和折射率难以与实验上的吸收分离,因此可以间接地分析散射对光路长度的影响。在作者以前的工作中,曾经描述了一种模拟确定单液相中光子通量和光路长度的实验方法。与单相流动的测定方法相比,化学光度法测定需要实验测定液体在反应器中的停留时间和液体在反应器中所占的体积。由于润湿相所形成的薄液膜的存在,气泡的横截面比液相的横截面减小,因此液体的电阻时间和液体体积有时比使用液体和气体流量计算的值高。KU Leuven大学的S. Kuhn课题组的研究为理解气体的存在如何影响气液泰勒流中的光反应提供了理论依据。这是通过分析光微反应器中的光子输运和流体动力学来实现的。具体而言,在光微反应器中,在不同的气相组分下,对气泡和塞子的长度和体积、膜厚、液体停留时间、每液体量的光子通量和光路长度进行了实验定量测定。然后,利用多区光化学反应模型预测了两相流中光化学反应的观测加速度,该模型考虑了反应器中的液体分布和液体接收的光子通量。此外,利用气体分数的函数预测了两相流中光路长度的值。该项研究于2020年发表在化学期刊ChemPhotoChem上(doi:10.1002/cptc.202000065)。【实验过程】一、光微反应器几何学本研究所用的光电抗器是在硼硅玻璃中刻蚀的蛇形通道。通道内径1 mm,长度69.9 cm。板式微反应器放置在距发绿光的光源2厘米处。光源包含84个LED,其排列与微通道的形状相匹配(见图2)。每个LED的电流(IF = 8毫安)由驱动板控制。该电流的最大发射波长在525 nm左右。用注射器泵将液体引入反应器中,用质量流量控制器将惰性气体氮气引入反应器中。气体和液体流在玻璃板上的T结处相遇。在本文研究的所有条件下,都得到了泰勒流。讨论了引入反应器的惰性气体的量,即气体分数βG,该值由气体流量除以气体和液体流量之和(QG+QL)计算得出:图2 荷兰Chemtrix光化学微反应器系统的示意图二、气体分数对光度计转化率的影响用化学光度法研究了气液泰勒流中的光子输运。这种方法涉及到光化学反应,其量子产率是已知的。在绿光照射下,作者采用了1,2-双 (2,4-二甲基-5-苯基-3噻吩基)全氟环戊烯从闭式向开式异构体的光异构化反应。这种光化学反应的量子产率随照射波长而变化,在480 nm处的量子产率在0.015,而 620 nm处的量子产率在0.027。通过测量光化学反应后封闭式二芳基乙烯 (DAECF) 浓度的衰减量,可以反算出达到试剂溶液的每液量光子通量。通过在不同的DAECF起始浓度和固定光强下进行一系列的化学光度计测量,作者发现,在单相流中,光路长度是可以确定的。与单相流相比,这种确定光路长度的方法在气液相反应中变得更有价值。由于多相流的非均匀性,在没有光线跟踪模拟工具的情况下,光路长度是难以估计的。因此,在绿光下不同起始浓度的DAECF溶液发生反应。通过一个在线的UV-Vis光谱仪在微反应器的出口进行检测。在不同气体分数下进行实验,分析了惰性气体对光化学反应的影响,实验结果如图3所示。图3 在不同气体分数、βG、总流量为1.3mL min-1时,光度计的转换与DAECF浓度在单相和双相流动中的起始点的关系正如预期的那样,通过增加DAECF的初始浓度,这种转化减少了。最大转化率不超过24%。有趣的是,无论DAECF的起始浓度是多少,观察值在气体分数的函数上都有明显的差异。例如,在最高浓度下,与单相流 (9) 相比,在βG = 0.88(18) 的两相流中得到了2倍高的转化率。将在12 × 10-4M的起始浓度和1.3mL min-1的总流量下测得的换算值与背景中讨论的换算值进行了比较。为了便于比较,在β = 0.5的惰性相比下,通过两相流中的转换,使转换归一化。如图4所示,我们的光度测量中发现的变化与Terao和Nakano等人在Paterni-Büchi光反应中获得的值相似。然而,在Tel mesani等人的工作中出现了随着气体含量的增加转化率也呈指数增长的情况。它们所观察到的反应加速度明显较高。这可能是由于它们的溶剂(乙酸乙酯对甲苯和己烷)、较小的管径 (0.8 mm对1 mm) 和所用反应混合物的不同光吸收特性的物理性质不同所致。图4 在分散相分数β = 0.5处得到的转化归一化。将实验结果与Terao等人报道的实验数据进行了比较为了计算每体积的光子通量和化学光度测量的光路长度,我们需要确定在微反应器通过时该液的发光时间。这是通过停留时间分布 (RTD) 实验实现的。三、单相和双相流动中的液体停留时间当假定微反应器的行为类似于塞流反应器时,液体停留时间tR,calc,在单和两相流动可直接由反应器容积 (V型反应器)和总流速计算,如下所示:表1 计算了不同流动条件下单相流动和双相流动的tR、calc和实验液体停留时间tR、exp为了验证这一假设,作者对液相和气液相进行了RTD分析。RTD实验包括注入示踪剂溶液,然后在反应器的入口和出口处测量浓度随时间的变化。如表1所示,单相在1.3mL min-1处的停留时间为25秒,这与基于塞流假设的计算值接近。在两相流中,气相分数在βG = 0.23和βG = 0.72之间,观察到相同的现象。当气体分数最高为βG = 0.88时,实验液体停留时间为28 s,高于25 s的计算值。本文采用轴向色散模型 (ADM) 对单相和双相流动中的轴向色散进行了定量研究。图5比较了1.3 mL min-1下不同气体馏分的RTD曲线。当βG = 0.88时,单相流和双相流的分布最广。在βG = 0.23的最低气体分数处发现了最窄的分布。图5 由轴向分散模型 (ADM) 得到的单相和双相流动的RTD曲线,总流速为1.3mL min-1这些观察结果与以前报告的观察结果一致。Kreutzer等人将示踪剂的扩散与气泡长度与气泡长度之间的比值进行了关联,而气泡长度与液体流量和气体流量之间的比值有关。气体含量最高时的显著脉冲展宽可以通过气泡周围滞留的长液膜与允许示踪剂进入许多随后的滞留物的短液塞之间的不完全质量交换来解释。因此,在βG = 0.88的实验条件下,这种回流能解释28 s的较长液体停留时间。在单相和最高气相组分中,增宽率最高,但在光化学反应中表现出两种相反的性质。因此,我们可以认为,当轴向色散影响液体停留时间时,光化学反应速率不受其影响。这一点在作者的系统中得到了证实,但轴向色散对其它类型的光化学反应可能有更大的影响。四、单相流和双相流中的光子通量利用RTD研究的停留时间,计算了每液体量的光子通量,I0:其中εavg为平均摩尔吸收系数 (8468M-1cm-1),c (0) 为DAE CF的初始浓度,c (t) 为DAE CF溶液的终浓度, l为液体中的平均光路长度,?avg为平均量子产率 (0.021 mol Einstein-1), t R, exp为反应器中的停留时间。εavg和?avg的加权平均值在480-620 nm之间图6 当总流量为1.3mL min-1时,在单相和双相流动中,光通量随液体体积的变化,即光度计起始浓度的变化表2 每液量的光子通量,I0;用化学光度法测定了不同流动条件下的光路长度。TP表示两相流,SP表示单相流由于光路长度l反应器是未知的,因此它与Matlab (Fitnlm)中的非线性回归模型进行了拟合。所获得的光子通量/液体体积,如图6所示。正如可以观察到的那样,在不同浓度下,在各个数据点之间仅发现了有限的变化 (<5%) 。由于数据点随机地分布在I0平均值附近,因此认为这种变化是一种实验误差。因此,先前用于确定单相流中的I0和lreactor而开发的方法成功的运用在两相流的实验中,结果列于表2。将两相流中的光反应器性能除以其在单相流中发现的值,通过增加气体的含量,使光反应器性能得到改善。两相流动的特征是,I0在βG = 0.88处与单相流动相比较增长了2倍。此外,表2列出了在单相和双相流动中锁门发现的光路长度的值。确定的光路长度表示光子在两相流的不同液体区域中的平均行进距离。通过增加βG = 0到βG = 0.88之间的气体分数,使光路长度从0.80 mm减小到0.56 mm。在βG = 0.88处,光路长度明显减小,表明反应主要发生在液膜中,而不是液塞中。这是首次在实验上定量的说明了气泡对泰勒流光路长度的影响。五、传质限制的研究泰勒流具有液体再循环的特点,它比单相流动更能促进径向混合。液塞的混合可与液塞的转数或再循环速率有关。作者通过在相同的气体分数βG = 0.55和两个额外的总流速0.56mLmin-1和0.82mLmin-1下进行化学半衰期放射线测量来研究再循环速率的影响。如表3所示,无论流速如何,均获得了每液体量和光路的类似光子通量。这表明本研究中所进行的光反应不受液塞内再循环速率的限制。此外,为了进一步检查所进行的光化学反应是否是光子或质量传输受限,我们在不同的光强下进行了光化学反应。光源的强度与通过LED的电流成正比。表3 光子通量与液量之比较,用化学光度法测定I0和lreactor,固定气相分数βG和不同总流量Q因此,我们比较了4mA/LED和8mA/LED的光电抗器性能。如果光化学反应在单相中的传质受限,则在βG = 0.88时,光化学反应在两相流中的传质则不应受限,如表4所示,I0在βG = 0.88的单相和双相流动下,得到的8 mA/LED值是其两倍。结果表明,在单相流和双相流中,光化学反应均处于受光子限制的反应状态,这是由于两种情况下I0之比相近,且无累加速率对光化学光度测定结果的影响。因此,在作者的研究中,液体混合并不能作为提高两相流转化率的一个方法。但是,存在的传质限制是每一个单独的光化学反应和实验条件所特有的,需要相应的验证。表4 光子通量与液量之比较, 在4 mA/LED和8 mA/LED 下用化学光度法测定I0与光路长度lreactor六、气液流动模式的表征如前文所讨论的,气泡长度和液塞长度以及气泡周围液膜的存在对光电抗器的性能有重要影响。因此,这些流动特性是通过流动成像来测量的。图7示出了微通道中的泰勒流动模式,其决定因素在于横截面面积,A。图7 微反应器中气液泰勒流动的示意图(非比例示意图)作者认为气泡的长度Lb是同一气泡的前和后之间的距离,而液塞的长度Ls是两个连续气泡的后和前之间的距离。此外,还假定在沟道壁上形成了厚度为δ f恒定的润湿液膜,并围绕着气泡和液塞。假定这种液膜处于静止状态,因此气泡通过一个缩小的横截面面积Ab。如下所示,这些假设允许根据气泡速度来计算膜厚。图8 在不同气体组分βG和a) 1.3 mL min-1, b) 0.82 mL min-1和c) 0.56mLmin-1的总流速下得到的气液流动图像图8示出了在不同气体分数及不同流速下获得的流动模式。正如所观察到的,在所有研究的条件下,气泡可以近似为半球面帽。测量气泡和弹塞长度的结果值如图9所示。最低的和最高的气体与其他条件相比,气液流动的不稳定性更高,这种条件对平均气泡和液塞长度的标准偏差更大。这些气泡和液塞长度的变化可能由注射器泵的脉动,气泡的形成和离开通道或通道的非均匀直径造成。在1.3mLmin-1时,随着气相分数从βG = 0.23增加到0.88,气泡长度从1.9mm增加到12.8mm,液塞长度从5 mm减少到1.4mm。当总和Lb+Ls不是常数时,不能直接比较这些数值,但可以用长度比Lb/Ls来分析不同的流动模式。如表5所示,Lb/Ls随着气体分数的增加而增加。因此,较高的Lb/Ls下I0越大,转换效果越好(见图3)。此外,通过对βG = 0.55和总流量分别为0.56、0.82和1.3mL min-1的流动模式的比较,得出总流量对气泡长度和液塞长度无明显影响的结论。这些恒定的气泡和弹塞长度解释了为什么在βG = 0.55时,无论总流量如何,在化学光度测量中结果类似的原因。图9 比较了不同气相组分下的平均气泡长度Lb、塞流长度Ls及其和Lb+Ls, βG,总流量为1.3mL min-1。此外,还给出了βG = 0.55的长度和0.82 mL min-1和0.56 mL min-1的总流速表5 用流动成像法测定研究的气液流动模式的特征下一个研究的方面是气泡速度,ub。两相流表面速度Utotal,计算公式为:实际上,由于滞流膜的存在,气泡的运动速度比U值要快。从连续性来看,气泡速度ub将大于通道中的总速度U,其因子为A/Ab:气泡的速度是使用两相流动的记录视频进行实验测定的。从表5可以看出,测得的气泡速度比两相速度高1.15-1.18。由A/Ab比值的实验值可以很容易地确定横截面积Ab。Ab还用于计算液膜厚度如下:其中R是反应器通道的半径,Rb是气泡的半径。如表5所示,所测定的膜厚在35 μm左右,并且在所有研究条件下几乎保持恒定。在作者的微反应器中发现的较厚的液膜可能是由几个因素引起的:(1)有机化合物的存在改变了己烷的物理性质。(二)表面活性剂痕迹的存在 (Marangoni效应)。(iii) 通道的粗糙度导致膜厚的恒定值,而膜厚的恒定值不随鼓泡速度而变化。七、每单位液体体积的光子通量的预测在这项工作中。 Lb/Ls增大了??0并因此改善了转化率。但是,这种长度比不能反映膜厚的影响。因此,作者建议将反应区内的反应堆空间划分为一个特定的区域 ??0和液体体积。图10比较了为单相流和双相流定义的不同反应区域。单相流动由连续液膜的一个区域组成,其体积为Vfilm和Vbulk。膜区是根据两相流中实验确定的膜厚来定义的。图10 模型中考虑的反应区的示意性表示。a) 单相流动中的液区:液膜和液区。 b) 两相流动中的液区:液膜、液体和气泡盖周围的液两相流由三个区域组成,这是因为气泡帽周围有额外的液体。这一带的特征是Vcap的液量。尽管图10仅示出了单位单元(一个气泡和一个液塞),但这些体积Vfilm、Vbulk和Vcaps表示分布在液体膜、体液和气泡盖周围的反应器中的总体积。假设在气泡中不发生光反应,在没有传质限制的情况下进行光度计测量,光散射对光反应器性能没有显著影响。其次,建立了一个简单的模型,该模型基于每个液量的光子通量和每个反应区的液量。用化学光度法测定的光子通量I(0,Vliquid)表示在液膜中和液塞中接收的光子通量之和:通过对膜、体和气泡盖周围的液体量进行求和,可以计算出反应器中在一定时间内的液体量。值得一提的是,当用气体碎片βG估算反应器中的总液量V液时,对所有流动条件下的总液量被低估。因此,使用流动成像实验测定了该模型中使用的V液体的值,并在图11中进行了说明。图11 反应器中的液量、Vliquid、Vfilm、Vbulk和Vcap周围的变化、气相分数β G的函数、总流速为1.3 mL min-1。此外,在总流速为0.82mL min-1和0.56mL min-1时, βG = 0.55随着气相分数的增加,气泡体积增大, Vliquid从βG = 0时的0.55mL线性下降到βG = 0.88时的0.14mL。这种下降是由于Vbulk下降所致,因为Vfilm和Vcap分别在0.08 mL和0.02 mL处几乎是恒定的。因此,在液体bulk中,每液量的光子通量I(0,bulk)可由曲线的斜率确定,如下方等式和图12所示。进而根据相关公式计算出I(0,film),与I(0,caps),计算结果如图6所示。图12 在1.3mL min-1的总流速下,测定从斜率和线性回归的截距的区域光子通量/液量表6 区域光子通量/液量,用多区光化学反应模型所测定的I(0,film), I(0,bulk)和I(0,caps)在比较表6中总结的模型的结果时可以看出,液膜区的特征是每液量的光子通量略高于cap周围的区域,但它比液塞高3.3倍。这种变化是可以解释的通过 (i) 光吸收引起的光通量梯度,或 (ii) 泊松波速度分布引起的速度梯度,作者认为光衰减是最可能的原因。同时作者预测了每液量的总光子通量。如图13所示,在预测值和实验值之间找到了很好的匹配。图13 实验与预测的每液量光子通量之比较该模型的建立是为了解释和预测随着气体含量的增加而增加的情况。但是,该模型的应用可以推广到两相光化学反应的优化。通过将各流动条件下的绝对光子通量值与单相值进行比较,作者发现两相流动具有较高的光子损失。当在反应器中引入惰性气相时,会被液体吸收的光子的一部分会在周围环境中丢失。此外,当反应器的一部分被惰性相占据时,在两相流中的生产效率将降低。从作者的模型出发,可以提出一种新的优化策略,通过在薄膜的高辐照区和体液的较暗区确定最佳液体量,使泰勒流动特性(即气泡和弹塞长度、膜厚)得到合理的设计。八、光路长度的预测如前所述,光路长度随气相分数βG的减小而减小。如果可以预测这种变化,那么在不同的气体含量下工作时就不需要进行光度测量。通过绘制图像发现 (1 -βG) lreactor随气相分数βG的变化呈线性关系,如图14所示。在单相流动条件下,截距值为单相光路长度1 SP = 0.797 mm。实验结果表明,斜率可定义为单相光路长度与经验系数f = 0.026 mm之和。图14 光路长度与气相分数βG之间的相关关系重新计算了两相流中的光路长度,结果如图15所示。关联式能较好地预测气液流动中的光路长度预测值与实验值的最大偏差在8.4%左右。图15 实验与预测光路长度的比较【结论和展望】1、 作者报告了惰性相可以加速泰勒流在微反应器中进行光化学反应的速率。这项工作旨在提供一个关于气液泰勒流中的光子输运和流体动力学如何影响光微反应器性能。2、 实验测定了在Chemtrix微反应器中各种气体组分的气泡和液塞长度、液体停留时间、每单位液量的光子通量和光路长度。气泡和气体长度与气相分数有很好的相关性,且与总流量成正比。RTD测量得到的停留时间与采用塞流假设的计算值接近。结果表明,气态组分影响的轴向分散不会影响光反应器的性能。3、 随着微反应器中气体的量的增加,每液体量的光子通量呈指数增长。与单相流动相比,在最高气相分数为0.88时,出现了2倍的上升。与单相流动相比,相同的流动参数具有更小的光路长度,比单相流动的光路长度小30%。4、 这一工作表明,通过研究光子输运和流体力学,可以对两相流的光反应器的理解有了显著的提高。此外,本研究中描述的方法为在广泛的流动条件下表征两相光反应器提供了参考。如果需要改进两相光反应器的能效和产能,所建立的模型可以为流型的优化设计提供指导。原文:Photon Transport and Hydrodynamics in Gas-Liquid Flows. Part 1: Characterization of Taylor Flow in a Photo Microreactor., 2020, ChemPhotoChem https://doi.org/10.1002/cptc.202000065【一正科技简介】作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:Photon Transport & Hydrodynamics in Gas-Liquid Flows_2020
应用实例
2021.12.06

【中山大学】采用荷兰Chemtrix微通道反应器进行连续流反应教学实验
背景介绍现代化工业发展以绿色可持续为前提,减少三废的排放,提高能量转换率,加快再生资源的开发。而传统的釜式反应由于传质传热的限制,实时在线监测不精准等缺点,难于满足现代工业发展的高要求,致使环境污染严重。连续流反应器术从根本上克服了传统釜式反应的不足,近年发展迅速。英国,美国等多数工业发达国家正在逐步将传统的釜式反应器转变为连续流反应器,而且许多著名企业已经成功应用连续流工艺进行生产,如辉瑞、惠氏、罗氏制药、诺华制药、帝斯曼集团,英国的阿斯利康公司等。目前该技术日趋成熟,未来化工行业的趋势,企业需要大量相关有连续流背景的技术人员,因此多所世界著名高校将连续流化学单独开设课程,如苏黎世联邦理工学院化工学院、卡迪夫大学、华盛顿大学、根特大学、美国东北大学化学院、普渡大学、赫尔大学、马斯特里赫特大学等。目前中山大学将微通道反应技术教学课程加到化学化工教学课程中(本科、研究生教学),旨在加强学生对连续流技术的理解和技术推广,配置多套实验室用途的微通道反应器+一套中试设备。学生分组进行微通道实验(4-5人一组,采用labtrix start进行教学方法手册中的试验)、老师操作中试生产设备(采用kiloflow做同一实验)。对比试验条件、结果和相关数据,可以验证微通道反应无放大效应,并且操作过程安全、自动化控制,让学生真正能体会到流动化学的魅力所在。图1 中山大学连续流教学现场图片图2 中山大学Kiloflow微通道中试反应器图片Labtrix Start连续流微反应器用于教学演示的优点Labtrix Start 连续流微反应器现已在多个高校用于教学演示,其配置包括:温度控制器,两个泵,一个反应器支架,多个反应器芯片, “Micro Reaction technology on Organic Synthesis”教科书一本,教学方法一套及流动化学计算软件一套。Labtrix Start 连续流微反应器用于教学演示可以有助于学生理解连续流动化学技术,培训学生操作连续流反应器技能,及培养学生开发新型流动化学反应的能力。Labtrix Start 连续流微反应器用于教学演示具有以下优点:设备体积小巧:如图3,系统共分为4部分,每个部分尺寸均约为20cm×10cm×10cm(长×宽×高)。图3 Labtrix Start连续流微反应器整套设备反应直观:反应器芯片是透明玻璃材质,流体流动及反应过程清晰可见。图4 随反应进行反应器芯片中液体颜色逐渐加深反应时间短:保留时间1.2 s-100min,每天可完成20-30组不同的实验条件筛选或动力学曲线绘制。图5 不同温度下的动力学曲线反应多样性:不同规格反应器芯片有2-5个试剂入口,可实现多步反应。图6 反应器芯片示意图应用条件范围广:系统适用于-20℃至+195℃,20bar压力下的反应条件,可安全演示危险反应及快速强放热反应,如硝化反应,叠氮化反应,丁基锂反应等。操作简单:实验过程中温度,压力设置可通过数字键盘或旋钮简单设置,而且Chemtrix.B.V公司配套有相关教学方法手册以及流动化学计算软件,可帮助学生快速理解转换泵速及停留时间的关系,提高实验效率。连续流反应器高校用户评论荷兰南方大学应用多套Labtrix Start 连续流反应器应用于微反应器技术实验教学和早期实验条件摸索。Gino van Strydonck教授说 “在南方大学,我们希望学生学习、接触最先进的实验技术、设备。我们建立了微反应器实验室,选用Labtrix Start用于微反应器实验教学和优化、筛选反应条件。 ”比利时根特大学实验室有一套Labtrix Start连续流反应器和一套KiloFlow微通道反应器,用于早期方法研究和放大实验。Stevens教授说:“是时候让学生接触连续流技术,并让他们实际操作,领悟连续流技术带来的变革,这样,他们一旦进入工厂,可以将新的技术应用于生产”。公司简介作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403
应用实例
2021.12.02
【Graz大学案例】连续流闪速化学用于瑞德西韦合成中的C-糖基化反应
【背景介绍】新冠大流行截止2021年1月1号已导致全球超过9400万例病例和200万例死亡。在寻找有效药中,由Gilead Sciences研发的瑞德西韦是美国食品药品监督管理局(FDA)和欧洲药品管理局(EMA)批准的第一种药物。据报道,仅在2020年第3季度,该药的销售量为8.73亿美元。迫于全球的形势,大幅提高该药的产量才能满足全球需求。瑞德西韦的生产工艺非常复杂,据报道,考虑到原材料的采购,首批产品需要9个月以上才能完成。而连续加工在简化工艺中可起着至关重要的作用。实际上,在这条路线中的某些步骤中,已经在工厂级生产规模使用了连续流技术。在安全性,非对映选择性,过程控制和时空产量方面提供了改进。可以预见,通过将其他合成步骤也转换为连续流模式,这些优点将得到补充,而其他优点也将得以实现。该药物的合成路线如图1所示,该路线始于卤化吡咯并嗪胺1 / 1a与苄基保护的D-核糖-1,4-内酯2 发生有机金属介导的C-糖基化,以提供关键中间体3。然后再需要五步来提供API 瑞德西韦。已有文献报道在室温下连续流中进行的Grignard反应,其停留时间少于一分钟。在没有添加剂的情况下,以47%收率得到了产品。图1 瑞德西韦的合成路线而另一方面,基于有机锂的反应可以使用价格更便宜且质量更高的杂芳基溴化物1代替其碘化物类似物作为有机金属前体。有文献报道使用1,2-双(氯二甲基硅基)乙烷(BCDSE)作为双齿硅基保护基,在N,N-二异丙基胺的存在下与有机锂试剂反应,尽管反应温度低(-78°C)且反应时间长,但该方法迄今为止提供了最高的收率(74%)。实际上,与较温和的格氏试剂相比,微反应器和连续处理技术更适用于处理有机锂试剂,因为可以极大程度的改善后者的传质和传热。闪速化学(flash chemistry)领域擅长于处理流动中异常快速的反应(反应过程最多几秒钟),并已在许多情况下得到证明,可以实现原本不可能或不切实际的化学反应。特别令人感兴趣的是闪速化学的可扩展方式,这种方式可以规避在药物制造开发过程中面临的的规模扩大问题。而连续流化学可以同时减轻在合成过程开始时因这一具有挑战性的转变而给供应链造成的负担。Graz大学的Kappe等人描述了通过闪速化学发展连续流的方法,使用7-溴吡咯烷-嗪胺1作为有机锂前体闪速合成糖基化吡咯烷嗪胺3。通过应用微反应器技术,工艺温度可以从-78oC显著提高到-30oC。此外,整个转化过程的反应时间可以大大减少到仅8 s。该结果最近发表在Org. Process Res. Dev. 期刊上(https://dx.doi.org/10.1021/acs.oprd.1c00024)。【实验方案】1. C-糖基化釜式反应研究7-溴吡咯并三嗪胺1与2,3,5-三-O-苄基-D-核糖内酯2的C-糖基化反应包括三个步骤。在第一步,伯胺1被硅基保护基团(例如BCDSE)保护,释放出2当量HCl,并在第二步被有机锂碱中和。需要第三当量的有机碱才能执行Li-Br交换并获得杂芳基锂1-Li。最后,对内酯2的亲核进攻产生所需的糖基化吡咯并三嗪3。最初,为了检查该工艺转换为连续流的潜在问题,作者首先开始了釜式反应研究。首先,原料1在已经报道的0.2 M反应浓度下是混悬液,因此必须将浓度减半以确保原料1的溶解度。另一个重要的问题是加入BCDSE后会形成固体,这可能会导致流动反应器内发生堵塞。先前对固体进行的表征表明,该沉淀并不是受保护的胺,因为在0 ppm左右没有信号(因为1H NMR光谱中该信号对应SiCH3)。作者认为该沉淀物是杂环1的质子化形式,当1被质子化后,上保护基反应不能发生。在环境温度下观察到固体形成需要约5 s,因此必须要在很短的时间内引入碱用以清除HCl。不过,在流动装置中可以很容易地实现并控制这种短的停留时间。在釜式反应中,作者发现缓慢添加n-BuLi可获得良好收率。假定副反应由于传热不良,导致容器中出现局部热点,传质不良,进而导致局部化学计量失衡。这两种作用都可能导致锂化中间体的分解,并随着反应规模的增加而放大。但是,在连续流中进行反应会增强传热和传质,因此有望以可扩展的方式进行有效的反应。杂芳基锂1-Li对内酯的亲核加成是面临另一个关键挑战。过去的研究表明内酯的α质子可被有机金属试剂迅速去质子化,导致锂化杂环1-Li猝灭。已经证明,当使用格氏试剂时,路易斯酸添加剂可以促进亲核进攻。然而,不溶性和稀有金属物质(NdCl3和四丁基氯化铵)增加了该工艺的成本和复杂性。有机锂物质由于其具有较高亲核性,因此比格氏试剂具有更高的亲核加成选择性。在先前开发的格氏试剂的工艺中,作者已经观察到核糖内酯加成物的开链中间体受到亲核攻击而产生的副产物。而有趣的是,使用有机锂方法时,作者并未观察到上述的副反应。这可能是由于杂芳基锂1-Li的亲核性增强,促进与内酯2的快速反应,而不是与亲电子性较高的酮中间体发生优先反应,或者可能是由于Mg抗衡离子可以使溶液中存在较高比例的开链酮形式。为了了解去质子化相对于Li-Br交换的相对速率,作者进行了釜式实验,其中一次添加了1当量n-BuLi。出人意料的是,在加入1当量正丁基锂后,已经交换了34%的溴化物,加入2当量后,交换了62%。这意味着在这些条件下,两个反应的速率没有显着差异。但是,为了避免形成的杂芳基锂发生内部淬灭,作者认为应将去质子化和Li-Br交换这两个过程分步进行。由于已知n-BuLi在去质子的同时可以快速进行Li-Br交换,作者试图先使用N,N-二异丙基氨基锂(LDA)去质子,而LDA可以避免在该阶段时就发生Li-Br交换,而继续反应一段时间后再加入n-BuLi来解决上述问题。2. 初始连续流过程根据上述这些观察结果,作者设计了一种连续流装置(图2),确保在添加LDA(防止形成盐)之前保证杂环1和BCDSE的反应时间,然后再添加n-BuLi(以实现锂-溴交换)。作者选择了连续流微反应器Ehrfeld,FlowPlate,Hastelloy C-22 /sapphire ,该反应器配备有小体积的混合板(TG混合器,内部容积为350μL,通道宽度为0.2-0.5 mm)。这有助于在可放大的系统中进行快速混合和有效的传热。图2初步的连续流结果将杂环1(THF中0.1 M)与BCDSE(THF中1.0 M)混合,通过在101μL反应器体积后与LDA(THF /己烷中1.0 M)相结合,成功实现了微反应器中的操作。然后,引入n-BuLi(己烷中1.6 M)形成杂芳基锂1-Li,将其用内酯2淬灭,得到所需的糖基化吡咯烷嗪胺3。在第一次尝试中,作者实现了54%的测定产率,同时包含21%的质子化杂环副产物1-H。尽管未观察到固体形成,但杂芳基溴1的溶解度远低于其碘化物类似物1a,因此必须使其浓度低至0.1M。然后进行流动实验以研究该设置中的反应速率。当所有流速乘以1.5(总停留时间= 5.3 s),反应收率不变,这意味着这些停留时间都足以使得每个步骤中能够完全反应。相反,将流率减半(总停留时间= 16 s)导致产品3的收率差很多(31%)。这意味着在低流量下较差的混合和传热会严重影响实验的效果。与内酯化步骤相比,内酯2上杂芳基锂1-Li的亲核加成预期进行得较慢,进而作者对停留时间的更详细研究。引入内酯2后,安装了毛细管反应器,可提供额外的38 s停留时间。当将毛细管反应器升温到-40或-20°C时,反应的产率与先前的结果一致,这表明亲核加成反应在微反应器板的最初5.3 s内已经完成。因此,额外的毛细管反应器被证明是不必要的(图2,条目4和5)。在连续流中,提高耐热性是改进有机锂反应的关键。虽然在釜式中糖基化过程需低温(-78°C),但在-50至-30°C之间的连续流装置中也得到了基本一致的结果(54-56%的收率)(图2,条目6和7)。进一步施加-20°C的高温会使产率降低到50%(条目8),因此选择了-30°C的最佳反应温度用于进一步的实验。3. 连续流工艺的进一步优化如作者最初的实验所示(图2),LDA充当强碱,可保持溴化物的稳定性(图3a)。因此,将LDA用于脱质子化,然后进行n-BuLi进行Li-Br交换被发现是最合适的组合。LDA筛选的化学计量在1到2当量之间(图3b)。增加LDA的量增加了产物3的产率,同时减少了质子化1-H的量。图3 杂芳基锂的内部淬灭及其通过改变LDA化学计量进行预防(a)由于快速的Li-Br交换而引起的内部淬灭至1-H的机理。(b)采用不同的LDA化学计量比。尝试改变去质子化和锂-卤素交换步骤的实验结果作者发现在糖基化过程中,形成的其他主要副产物是锂化杂环1-Li对硅基保护基团的亲核攻击所致(图4a)。中间体5继续与有机锂反应,或在淬灭/ HPLC分析期间简单地水解,形成副产物6-8。作者认为,N-Si键(与C-Si键不同)在水的存在下是不稳定的,因此也可能存在其他无法检测到的中间物种。图4杂芳基锂与硅基氯的副反应(a)作者观察到的杂芳基锂与BCDSE反应产生的副产物,根据LCMS分析提出了结构。(b)筛选BCDSE化学计量以最大程度减少副产物6-8形成这些副产物的存在暗示了BCDSE保护基可能存在不完全消耗的问题。为了解决这个问题,作者将BCDSE的化学计量从1.0当量逐渐降低到0.5当量(图4b)。出人意料的是,总收率未受到明显影响,并且硅基的副产物的数量减少。为了便于纯化(消除硅基的副产物)和降低成本,作者决定使用0.6当量进行操作。由于即使使用亚化学计量的BCDSE也可以实现稳定的流动过程,因此作者在不存在任何保护基的情况下,分批和流动地进行了实验。使人惊讶的是,釜式反应的产率为20%,流动化反应的产率为49%。不幸的是,在没有硅基保护基的情况下,1与n-BuLi的反应几乎立即形成了固体,这使得该流动过程不可能长时间运行。4. 过程稳定性实验在对反应参数进行其他精细调整之后,作者已经开发出产品达到60%分离收率的反应条件,总停留时间仅为8 s(图5a)。为了证明该过程在更长的处理时间内的稳定性,作者进行了2小时的长时间运行,每5分钟进行一次采样(图5b)。实验结果表明该过程是非常稳定,在整个操作期间,产率仅变化图5优化的反应条件和长期稳定性的连续流装置的示意图(a)EhrfeldFlowPlate(TG混合器)的示意图以及反应条件。(b)长期运行期间随时间变化的反应的结果收集反应产物,得到16.9g(收率60%,纯度96%)糖基化产物3。在这种情况下,该材料已通过柱色谱法纯化,但在放大规模中可以使用现有的结晶方法。这相当于8.5 g/h的通量,以及极高的时空产率10.4 kg L-1 h-1。高时空产率清楚地表明了采用闪速化学方法进行这些转化的优势,即使在小型反应器中,较短的反应时间也可以实现高通量。当与放大规模反应相结合时,将这种成熟的反应连续流平台升级到更大的版本,可节省大量的成本,时间和能源。与报道的使用NdCl3和TBACl添加剂的格氏试剂的间歇方法相比,该方法的优点在于所需添加剂的成本较低。虽然BCDSE是一个比较奇特的硅基保护基,但对每摩尔1a的加工成本仅为179欧元。与之相对的是,所报告的釜式工艺中每摩尔1a的加工成本为2091欧元。当然,必须指出的是,这些价格并不代表制造规模下的结果,但是随着对无水NdCl3的需求的增多可能会增加大量成本。【实验结论】作者探讨了基于有机锂的C-糖基化连续流合成瑞德西韦的工艺,通过仔细分析反应混合物和添加顺序,可以避免在微反应器内形成固体。又通过对反应参数的优化,包括硅基保护基和有机金属试剂的筛选,使得转化中副产物的含量降至最低。作者通过精确调整工艺参数,再加上微反应器中赋予的优异的传热和传质能力,可在仅8 s的总停留时间内进行高度放热的C-糖基化反应。与釜式反应所需的-78°C相比,连续工艺在-30°C的更高的过程温度下,该反应得以成功实现,这将节省大量能源。已建立并开发的连续流可扩展的反应器系统,具有实现更有效地生产关键糖基化中间体和相应API的潜力。原文:Flash Chemistry Approach to Organometallic C?Glycosylation for the Synthesis of Remdesivir, 2020, Org. Process Res. Dev(https://dx.doi.org/10.1021/acs.oprd.1c00024)【公司简介】作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20210329/20210329170417_7085.pdf
应用实例
2021.12.02

【复旦大学案例】 基于不对称Henry反应实现酰胺醇类抗生素--氯霉素、叠氮氯霉素、甲砜霉素、氟苯尼考的连续流合成
【背景介绍】酰胺醇类抗生素是一类重要的合成抗生素,对革兰氏阴性和革兰氏阳性微生物(如链球菌属、葡萄球菌属和巴氏杆菌属)具有广谱抗菌活性。1(–)-氯霉素(1),(–)-叠氮霉素(2),(+)-甲砜霉素(3)和(+)-氟苯尼考(4)(图1)是该抗生素家族的代表性药物。2它们已被有效地用于治疗人类和活牲畜的过敏性和严重细菌感染。由于此类抗生素在全球范围内使用量巨大,如何简捷高效合成此类抗生素,具有重要的实际意义。[图1. 酰胺醇类抗生素(1-4)的结构]通过逆合成分析(图2),我们发现2-氨基-1,3-二醇(6)为合成酰胺醇类抗生素(1-4)的关键中间体。该中间体含有两个相邻的手性中心,因此,大规模生产此类高光学纯2-氨基-1,3-二醇(6)仍具挑战。中间体2-氨基-1,3-二醇(6)可以通过催化氢化2-硝基-1,3-二醇(7)中的硝基获得。高光学纯2-硝基-1,3-二醇类化合物(7)可以通过芳香醛(8)与硝基乙醇(9)的不对称Henry反应制备。[图2. 逆合成分析]此合成路线涉及催化氢化,传统釜式氢化工艺属于高危工艺,氢气易燃易爆,而催化加氢往往需要在高压下进行,对设备、工艺、安全、人员的要求非常高。近年来,连续流技术发展日趋成熟,与传统釜式反应技术相比,连续流有诸多明显优势:1)传质传热效率是传统釜式反应的上千倍,极大的缩短反应时间;2)单位反应体积小,反应安全性大大提高;3)物料配比精确,反应条件精确控制,生产重现性好;4)工艺过程连续不间断,自动化程度高。3基于以上优点,作者在后续反应中引入连续流技术,突破传统反应,高效快速合成酰胺醇类抗生素(1-4)。[图3. 酰胺醇类抗生素(1-4)的合成路线]四川大学华西药学院的陈芬儿院士课题组报道了甲砜基苯甲醛与硝基乙醇的顺式选择性不对称Henry反应,成功构建一系列高光学纯的2-硝基-1,3-二醇类化合物(图3)。随后,作者通过对硝基进行连续氢化获得酰胺醇类抗生素的关键中间体2-氨基-1,3-二醇类化合物。最后,作者通过多步连续流过程,高效快速的完成了四种酰胺醇类药物的不对称合成。文章发表在 J. Org. Chem. 2021, 86, 17, 11557–11570(https://doi.org/10.1021/acs.joc.1c01124)。【实验方法】作者以甲砜基苯甲醛(8b)和硝基乙醇(9)为标准底物,以10 mol %一水合醋酸铜和氨基醇配体L1的络合物为催化剂,四氢呋喃为溶剂,室温反应24小时后,能以68%的收率,中等的非对映选择性(dr = 73:27)和非常高的立体选择性(97% ee)得到目标产物(表1)。随后,作者考察不同种类的氨基醇配体,发现联苯类氨基醇配体L9,为最优配体。虽然,收率有所提高(80%),但非对映选择性仅仅略有增加(dr = 80:20)。接下来,考察其他种类的反应溶剂,如乙腈、二氯甲烷、1,2-二氯乙烷和N,N-二甲基甲酰胺,发现四氢呋喃仍是最优溶剂。降低反应温度到0℃时,该反应的立体选择性可以极大提高(dr = 97:3, 97% ee),但收率较低(23%)。高兴的是,增加硝基乙醇的用量、提高反应浓度和延长反应时间收率可以显著提高(98%)并且保持非常高的立体选择性(dr = 97:3, 97% ee)。降低催化剂的用量到5 mol %,该反应也能保持极好的化学反应活性和立体选择性,但是需要大大增加反应时间。[表1. 不对称Henry反应的条件优化a]a 反应条件为:8b (0.2 mmol), 9 (0.6 mmol), Cu(OAc)2.H2O (10 mol %) 和 L (10 mol %) 在0.8mL溶剂中,25 °C反应温度下反应24小时。b 分离收率。 c顺反比和ee值通过手性HPLC测定。 d 反应温度为0 °C. e 反应温度为-5 °C。 f 9 (4.0 equiv) 溶于0.3 mL 四氢呋喃。 g Cu(OAc)2.H2O (5 mol %), L9 (5 mol %)。在获得最优反应条件之后,作者又对芳香醛底物的适用性进行了考察(表2)。在苯环上不管是引入吸电子基还是给电子基,该顺式选择性的不对称Henry反应都可以顺利进行并且能以良好的收率和极好的立体选择性得到顺式2-硝基-1,3-二醇类化合物(7a-7t)。此外,多取代苯甲醛类底物(8u-8x)也能很好的兼容该反应体系,尤其是三甲氧基取代的苯甲醛(8x)为底物时,非对映选择性高达99:1以及大于99%的对映选择性。另外,萘醛(8y)和芳香杂环醛(8z和8aa)也可以很好的适应该催化体系。[表2. 芳香醛底物的适用性考察a]a反应条件: 芳香醛8 (1.0 mmol),硝基乙醇9 (4.0 mmol), Cu(OAc)2.H2O (10 mol %)和L9 (10 mol %) 溶于1.5 mL的四氢呋喃并在0 °C下反应72-120 小时。收率为分离收率。 反应顺反比和ee值通过手性HPLC检测。b 转化率通过HPLC检测。在确定了顺式2-硝基-1,3-二醇类化合物7a (95% ee) 和 7b (97% ee) 的合成方法之后,作者开始尝试连续合成酰胺醇类抗生素 (1-4) (图4)。首先是氯霉素(1)和叠氮氯霉素(2)的连续合成。近年来,微通道连续氢化反应多有报道。4在此基础上,作者将10% Pd(OH)2/C分散在石英砂,然后填充于深圳市一正科技有限公司的微通道固定床反应器(MF-200)中并测得该反应器保留体积为5.0 mL。[微通道固定床反应器]随后作者将2-硝基-1,3-二醇类化合物7a溶于甲醇中并用柱塞泵以0.5 mL/min的流速泵入盘管式混合器中。在混合器中,气液两相进行混合,其中氢气流量通过气体流量计控制在0.1 L/min。随后,反应液进入固定床微反应器中,进行气液固三相加氢反应,保留时间为10 min。通过LC-MS检测,2-硝基-1,3-二醇类化合物7a的转化率为99%。从对硝基苯甲醛(8a)到2-氨基-1,3-二醇(6a)两步总收率为76%。作者发现,填充于固定床中的催化剂在室温下可稳定存在十天以上并且保持其催化活性。随后,作者开展连续酰胺化反应的研究。将2-氨基-1,3-二醇(6a)和二氯乙酸甲酯(10a)分别溶于甲醇中,通过两台注射泵,分别注入T形混合器中进行混合。随后混合液通入内径为0.8 mm, 体积为1.0 mL的PTFE材质的盘管反应器中,在反应温度40℃,背压5 bar,保留时间为3.5 min的条件下,能以80%的分离收率得到酰化产物5a。在相同设备和反应条件下,2-叠氮乙酸甲酯(10b)也可顺利参与连续酰化反应,以81%的分离收率得到酰化产物5b。最终,作者通过对芳香胺的连续氧化得到氯霉素(1)和叠氮氯霉素(2)。酰化底物(5)溶于丙酮和水的混合溶剂,通过碳酸氢钠和氢氧化钠缓冲体系维持反应液为弱碱性(pH = 7-8)。氧化剂Oxone?溶于EDTA水溶液中。酰化底物(5)溶液和Oxone?溶液分别通过注射泵泵入T形混合器,接着,先在0℃下通入盘管反应器(保留时间为45秒)。然后再25℃下通入另一盘管反应器(保留时间为45秒)。在此反应条件下,氯霉素分离收率为62%,叠氮氯霉素分离收率为64%。随后,作者又开始连续合成甲砜霉素和氟苯尼考。粗品2-硝基-1,3-二醇类化合物7b的甲醇溶液通入相同的固定床反应器,进行连续氢化,以82%的收率得到2-氨基-1,3-二醇6b。化合物6b与二氯乙酸甲酯在盘管反应器中进行连续酰胺化反应,即可以84%的分离收率得到甲砜霉素。化合物6b同样也是合成氟苯尼考的关键中间体。化合物6b与二氯乙腈在盘管反应器中,停留时间仅10 min, 便能以88%的收率得到环合中间体11。随后,中间体11与Ishikawa’s试剂在盘管反应器中进行连续氟化反应。在二氯甲烷溶剂中,反应温度为100℃和10 bar背压条件下,2.5 min便能以100%的转化率得到氟化中间体12。随后,氟化中间体12在异丙醇与水的混合溶剂中进行连续开环反应,最终,以81%的收率(两步)得到目标产物氟苯尼考。[图4. 连续合成酰胺醇类抗生素(1-4)]【结论】1) 作者通过不对称Henry反应高立体选择性和化学选择性合成一系列顺式手性2-硝基-1,3-二醇类化合物并将其应用到酰胺醇类抗生素(氯霉素、叠氮氯霉素、甲砜霉素、氟苯尼考)的合成。目前为止,此合成路线最短,收率最高:氯霉素38%的总收率(4步);叠氮氯霉素39%的总收率(4步);甲砜霉素69%的总收率(3步);氟苯尼考58%的总收率(5步)。2) 作者运用微通道连续加氢技术代替传统釜式加氢反应,大大缩短反应,提高反应效率。催化剂无需回收,可连续使用10天以上并保持其催化活性。3) 基于连续流技术的优势,作者将后续釜式反应改变为连续流反应,大大提高了此类抗生素的合成效率,为以后的放大生产及工业化生产打下了坚实的基础。【原文】J. Org. Chem. 2021, 86, 17, 11557–11570The Journal of Organic Chemistry (https://doi.org/10.1021/acs.joc.1c01124)【一正科技简介】作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20211201/20211201153422_7399.pdf/include/upload/kind/file/20211201/20211201153444_3841.pdf
应用实例
2021.12.02
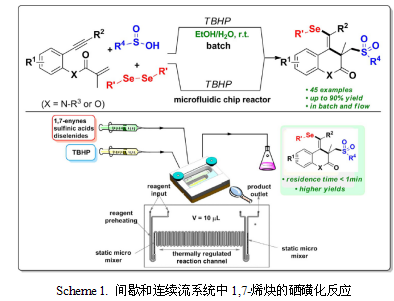
【南京工业大学案例】间歇及连续流系统中无需金属自由基引发的1,7-烯炔硒磺化反应
【摘要】以1,7-烯炔为原料,经亚磺酸和二苯基二硒醚硒磺化在间歇及连续流系统中制备多功能3,4-二氢喹啉-2(1H)-酮。该室温方案提供了一种可以采用多种底物来制备硒代砜的方法,反应产率中等甚至优异。这将为医药和生物化学领域构建多种有意义的3,4-二氢喹啉-2(1H)-酮的衍生物提供一种新方法。此外,间歇式反应需要20 h,采用荷兰Chemtrix微通道反应器Labtrix Start,反应明显加速,缩短至43 s。【前言】有机硒砜是一类极其重要的化合物,在药物化学和有机合成领域有着广泛的应用。砜和硒官能团可以通过多种方法从目标分子中引入和去除,因此它们也被广泛用作合成各种天然产物的关键前驱体。因此,一锅法硒化反应来制备硒砜化合物激起了相当大的研究兴趣。例如,Zhang等人开发了一种铜催化的炔烃硒磺化反应来合成(E)-β-硒乙烯基砜;Liu等人利用芳基磺酰肼和二苯基二硒化物实现了乙炔的硒磺化。尽管已取得显著进展,但合成3,4-二氢喹啉-2(1H)-酮化合物的1,7-烯炔自由基引发的硒化反应尚未被报道。另外,氧化自由基1,7-烯炔环化反应是一个构造各种复杂环形分子的有效方法。这类反应通常有着理想的环化效率和功能兼容性,且不需要底物的预功能化及过渡金属的使用。近年来,业内将重点放在了1,7-烯炔自由基环化反应来构建多功能碳环和杂环骨架上。受这些结果的启发,我们预测:在适当的条件下,亚磺酸原位生成的磺酰自由基更倾向于参与1,7-烯炔末端的双键加成反应,并被其他自由基捕获生成新的功能杂环分子。正如预期,1,7-烯炔与亚磺酸及二硒化物成功发生磺酰自由基引发的环化反应,生成3,4-二氢喹啉-2(1H)-酮,收率优良。在此,我们提出了这种新型的包含两种自由基的三组分一锅法硒化反应。该反应在间歇和连续流系统中都可有效地发生,并实现中等以上的收率(Scheme 1)。【结果与讨论】首先选取N-[2-(苯乙炔基)-苯基]-N-对甲苯磺酰甲基丙烯酰胺(1aa, 0.2 mmol)、4-甲苯亚磺酸(2a, 0.6 mmol)及二苯基二硒化物(3a, 0.1 mmol)为模型反应物来优化条件(Table 1)。该模型反应在EtOH中及空气条件下进行。室温下以H2O2 (3.0 equiv)为氧化剂,可以得到预期产物5aa,但收率仅为20%。随后尝试利用其他氧化剂来提高转化效率,如二叔丁基过氧化物(DTBP)、叔丁基过氧化苯甲酸酯(TBPB)、1,3-二氯-2-丙醇(DCP)、过硫酸钾(K2S2O8)、叔丁基过氧化氢(TBHP)、3-氯过氧化苯甲酸(m-CPBA)和次氯酸钠(NaClO)。结果表明,TBHP的性能最佳,能以72%的收率得到硒化产物5aa。此外还对各种溶剂进行了筛选,包括1,4-二氧六环、二氯甲烷(DCM)、丙酮、乙酸乙酯(EA)、甲醇(MeOH)、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)、H2O和EtOH/H2O。结果发现,EtOH/H2O (5:1 v/v)的共溶剂体系为该反应的最优溶剂条件,然后将温度提高到40 ℃,5aa的产率为76%。进一步提高反应温度,产率则明显降低。然后通过使用0.2 mol%的不同催化剂,包括I2、四丁基碘化铵(TBAI)和FeCl3,并改变底物比及其它条件来进行优化反应。研究发现:在没有任何催化剂的情况下,1:3:0.75 (1aa:2a:3a)的底物比及250 ℃的温度条件可以实现最高的5aa产率,为88%。氧化剂(3.0当量)、溶剂(2 mL)置于密封的反应管中,在空气中放置20 h。在确定了1,7-烯炔的硒磺化最佳反应条件后,我们尝试利用不同N-取代1,7-烯炔、亚磺酸和二硒化物来进行硒磺化反应。如Scheme 2所示,首先以对甲基亚磺酸2a和二苯基二硒化物3a为磺酰自由基供体和硒源,探索1,7-烯炔上取代基(R1-R3)对反应的影响。结果表明,当R3位置为Ts基团时,炔基基团(R2)上的各种乏电子或富电子的取代基都对这一自由基诱导过程没有影响,从而使5aa和5ag有着中等到优良的产率。其中,当对甲氧基苯基(PMP)对应物(1ac)为反应物时,反应收率略有提高(5ac, 90%)。而叔丁基的存在又会导致产率的显著降低(5ae)。再者正丁基对应物1af并不是这个反应(5af)的合适底物,这可能是由于过程中原位生成的乙烯基自由基中间体相对不稳定造成的。此外,无论苯磺酰保护基团的对位取代基的电子特性如何,都可以实现高的反应产率(5ba-5bl)。对不同取代模式和电子特性的亚磺酸进行测试。亚磺酸2的苯环上既具有富电性基团(MeO, 5cc)、中性基团(H,5ca;甲基,5cb;t-Bu, 5ch),也具有缺电性的(Cl, Br, F, CF3, 5cd-5cg)基团。结果表明,磺酰基自由基引发的内成键6元环化反应成功发生,N-磺酰化喹啉-2(1H)-酮5ca5ck的产率为65~86%。且底物2苯环上的供电子取代基比缺电子取代基更有利于反应发生。而且幸运的是,带有其他磺酰基如2-萘基、2噻唑和环丙基的底物2也适用于这个过程(5ci-5ck)。还探索了1,7-烯炔的内部芳烃环的4位或5位上的官能团(R1) 对反应的影响,包括甲基、甲氧基、溴和氟。结果表明,这些取代基1da-1de均可得到目标产物5da-5de,收率为62%~86%。此外,二甲基二硒化物也适用于该转化反应,可以80%的产率制得硒磺化产物5fa。最后通过X射线分析确认了5aa的结构。密封反应中的氧化剂(3.0当量)、溶剂(2 mL)在空气中放置20小时。有趣的是,在标准条件下各种O-取代的1,7-烯炔4a-4d都可以硒磺化反应,得到苯并二氢吡喃-2-酮衍生物6a-6d,产率为46-70% (Scheme 3)。可见,与N-取代的1,7-烯炔相比,O-取代的1,7-烯炔的反应转化率相对较低。这是由于氧原子的电负性大于氮原子的电负性,从而降低了底物的反应活性。为探究其机理,我们进行了对照实验(Scheme 4)。在自由基清除剂TEMPO(2,2,6,6-四甲基哌啶1-氧基)或BHT(2,6-二叔丁基-4-甲基苯酚)的存在下,1,7-烯炔1aa与2a和3a在标准条件下发生反应,但未观察到产物5aa。这表明该反应为SET型机理(Scheme 4a)。而如果没有氧化剂,该反应不会发生,底物可完全回收(Scheme 4b)。在不含亚磺酸的情况下,标准条件下处理1aa和2.0当量的二苯基二硒化物3时,未观察到预期产物7 (Scheme 4c)。最后,以4-甲基苯磺酰肼(1d)为磺酰源,标准条件下5aa的产率为82%( Scheme 4d)。这些结果证明由芳基亚磺酸原位生成的芳基磺酰基自由基可以有利地引发加成环化反应,形成乙烯基中间体,然后截取苯硒基自由基。因此,我们可以推测,磺酰化发生在硒化步骤之前。根据本文研究结果以及业内相关报道,Scheme 5提出了反应机理。首先,芳基亚磺酸在TBHP的作用下经过单电子转移过程氧化形成磺酰自由基A。类似地,二苯基二硒化物也生成苯基硒基自由基。随后,磺酰基自由基A攻击1,7-烯炔1aa的末端烯烃结构,生成自由基B,进而发生6-外-dig-环化反应,形成乙烯基自由基中间体C。在苯硒酰基自由基的存在下,中间体C通过自由基偶联转化为最终的3,4-二氢喹啉-2(1H)-酮5(Scheme 5)。我们目前正在进行进一步研究来揭示更详细的反应机理。与间歇反应相比,连续流反应有许多优势,特别是在自由基化学领域。所以为了提高反应效率,缩短反应时间,我们尝试将1.7-烯炔1的三组分间歇反应换为连续流反应。我们采用了荷兰Chemtrix微反应器Labtrix Start流动系统。它由一个微流控芯片反应器和两个注射器泵组装而成。微流控芯片反应器和注射器的体积分别为10 μL和1000 μL。通过改变注射器的流量,可以改变反应物的摩尔比和反应时间。最终在优化相对流速和溶剂条件后,5aa的收率达到了90% (Table 2)。尤其当流速为7.0 μL/min时,停留时间仅为43 s。产品收率的提高(连续流的90% vs间歇反应的88%)及反应时间的缩短(连续流的43 s vs间歇反应的20 h)可以解释为微流控反应器的较短的长度尺度带来了高的反应效率。该反应条件同样也适用于其他硒砜类化合物。比如在相似的反应条件下,5ba和5ca的产率分别高达85%和86%。此外计算结果表明,微流控芯片反应器中的时空产率要显著高于间歇釜的时空产率。mily:Times;color:rgb(0,0,0);font-size:10.5000pt; mso-font-kerning:1.0000pt;" >L和1000 μL。通过改变注射器的流量,可以改变反应物的摩尔比和反应时间。最终在优化相对流速和溶剂条件后,5aa的收率达到了90% (Table 2)。尤其当流速为7.0 μL/min时,停留时间仅为43 s。【结论】综上所述,我们开发了一种新型三组分硒磺化方法,以实现1.7-烯炔的内成键6元环化反应。该方法无需金属自由基的引发,在间歇和连续流系统中,成功合成了多种多种官能团的3.4-二氢喹啉2(1h)-酮化合物。连续流系统将反应时间从20 h缩短到43 s,表明了连续流动系统的突出优势。目前正在进行进一步研究以揭示这类反应的机理,并探索在其他硒砜化合物合成中的应用。【参考文献】[1]Metal-free radical-triggered selenosulfonation of 1,7-enynes for the rapid synthesis of 3,4-dihydroquinolin-2(1H)-ones in batch and flow[J]. Advanced Synthesis & Catalysis, 2017, 359.【一正科技简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20210823/20210823172559_9607.pdf
新品
2021.12.01

【华东理工大学案例】采用Conflore ACR连续多级搅拌反应器连续流合成聚合物改性添加剂的重要前体——解决了沉淀反应在连续流中的稳定运行问题
【背景介绍】2,2’-亚甲基-双(4,6-二-叔丁基苯氧基)磷酰氯(2)是NA-40,NA-45以及NA-71等一系列聚合物成核剂的合成前体,在精细化工中占有重要地位。通常,它由2,2’-亚甲基-双(4,6-二-叔丁基)苯酚(1)和POCl3在三乙胺作为缚酸剂的条件下经磷酰化反应制备(溶剂为甲苯,常压,产物经水解处理后用于表征)。同时生成固体沉淀产物三乙胺盐酸盐(3),如图1所示。虽然在间歇式反应器中反应收率已经优化至90%,然而此过程中仍然存在传热效果差、生产效率低等显而易见的缺陷。图1近年来随着连续流技术在合成领域逐渐崭露头角,其良好的传质传热性能,精确的过程控制能力以及出色的工艺安全性为解决上述问题提供了潜在可行的方案。然而,由于反应产物三乙胺盐酸盐(3)以固相沉淀的方式逐渐产生,因此反应过程中极易造成连续流设备的不稳定操作甚至最终堵塞,而这一问题也一直是连续流反应器功能适用性的一项严峻挑战。针对这一棘手问题,华东理工大学辛忠团队设计了几种不同形式的连续流反应系统并验证了它们对这一有机沉淀反应的耐受性。实践证明,一种特制的连续搅拌反应器Coflore ACR在处理含固多相反应体系时表现极为出色。基于此,本文作者第一次在连续流反应器中合成了这一重要的聚合物成核剂前体,并优化了反应工艺使收率达到了98%。文章发表在2021,Org. Process Res. Dev(https://doi.org/10.1021/acs.oprd.1c00105)。【实验部分】在进行连续流实验之前,作者曾设想是否可以先在间歇反应器中通过改变工艺条件(如溶剂、缚酸剂、催化剂的种类以及温度等)来“消除”沉淀产物,同时还能将反应收率优化至更高。如此连续流反应器将不会面临堵塞的风险。然而一系列的优化实验证明,“甲苯+三乙胺”仍然是兼顾各个考量因素后的最优选择,因此处理沉淀产物需要依靠其它方式。基于全氟烷氧基树脂(PFA)材质的盘管式连续流反应器是形式最简单且最常用的连续流反应器种类之一。作者将PFA盘管反应器置于超声水浴中,期望强烈的声波场可以阻止沉淀颗粒在管壁上的黏附效应,相应的工艺流程图如图2所示。图2然而实验结果表明,即使施加强烈的超声外力场,连续流反应器的稳定运行时间一般也难以超过30分钟,相比于无超声环境,运行时间也就仅仅延长了一半左右。而若想实现长时间的规模化生产,这一时间尺度显然是微不足道的。这意味着沉淀产物的积聚只是被延缓而并未被消除,堵塞最终还是会发生。虽然降低流速、反应物浓度、温度并扩大管径也能在一定程度上延缓堵塞的发生,但终究不是长久之计。不过,“对反应系统施加外力场(机械能)”这一思想为作者解决这一问题打开了新思路。搅拌通常被认为是处理含固多相反应体系的有效手段,而由此发展出的“连续搅拌反应(CSTR)”原理已经被认为是在连续流中处理多相反应体系应用最广泛且最有效的方法。基于此,作者自主设计并评价了一种连续搅拌管式反应器(CVTR):材质为316不锈钢,长4.5米,内径4.35 mm,壁厚1 mm,内含1500个直径3 mm的316不锈钢珠作为搅拌子,反应体积为45 mL,实验流程图如图3(a)所示。将反应器置于一个可以提供50 Hz定频、5 mm振幅的可持续垂直振动的振动台上,钢珠在振动作用下可以对含固的多相流动物料进行充分的搅拌和剪切。因此,整个反应器相当于一个多级串联的微型CSTR,如图3(b)所示。图3为便于与相同材质和体积的空管式反应器进行性能比较,作者定义了当量半径Re,意义是具有与CVTR同样长度和反应体积的空管的半径,经计算为1.8 mm。因此,作者比较了CVTR和空管式连续流反应器(316不锈钢材质,长4.5米,内径4 mm,壁厚1 mm)对沉淀反应的耐受性。实验结果表明, 空管式反应器在30 min左右即被反应沉淀所堵塞,而CVTR可实现明显更久的稳定运行,从而验证了CVTR处理沉淀反应的优良性能。然而,一旦停止振动,堵塞很快就会发生,而长期高频的机械振动又会对反应系统造成不可避免的机械松动和磨损,以至于外部换热夹套都无法有效安装。因此,反应只能在室温(30 ℃)下进行,收率也只有75.9%。虽然延长停留时间可以进一步提高收率,但也大大降低了反应通量。而反应物POCl3作为强酸性物质,势必会对反应系统中的不锈钢材质进行缓慢腐蚀,这也进一步表明了CVTR自身的局限性。图4图5首先研究了振动频率对反应收率的影响。实验结果如图6(a)所示,可以看出,振动频率对收率的影响不大,说明所测试的振动频率已经对物料提供了足够强烈的混合,重点在于防止沉淀的积聚,反而当频率降低到4或5 Hz时,反应系统压力的缓慢上升预示着堵塞的风险。兼顾性能和能耗,选用6 Hz的振动频率做进一步工艺优化。虽然进料方式采用的是底部两股分别进料,但仍然探究了预混合是否对反应收率有影响。将进料方式改为进料口前置T型混合器的预混合单股进料,并与两股进料进行对比,结果发现二者收率并没有什么区别,这说明反应器本身已经对物料提供了足够强烈的混合。况且,T型混合器和进料口前必须用一小段PFA管连接,而由于反应速率较快,两股物料接触即开始产生沉淀,最终快速积累的沉淀会堵塞PFA连接管。因此,不需要在本工艺中设置预混合。图6图7而后探究了反应温度和停留时间对收率的影响,结果如图7所示。可以看出,二者均对收率有明显的正向影响,且温度越低,影响越大。因此,提高反应温度可以弥补停留时间缩短引起的收率下降。在精细化工中,为了达到较高的反应收率和生产通量,提高温度所带来的能耗代价往往是次要的。为此,选用“4 min停留时间+100 ℃”来进一步优化工艺。通常,反应物料的配比对反应结果有明显影响。因此,首先探究了缚酸剂三乙胺与反应物酚(1)的摩尔比对收率的影响,结果如图8(a)所示。可以看出,当三乙胺当量从2.8下降至2.1,收率从97.6%剧烈下降至71%,对应反应混合物的pH也从7.0降至约3.0,说明此反应对碱性环境的强弱比较敏感。而从图9所示的反应机理以及间歇反应器中的实验结果也可以看出这一点。当三乙胺当量从2.8升至3.2,虽然pH继续上升,但收率反而从97.6%下降至90.3%,这可能是由于多余的三乙胺消耗了反应物POCl3。因此,三乙胺当量采用2.8为宜。图8图9最后探究了POCl3当量对反应结果的影响,结果如图8(b)所示。可以看出,当POCl3当量从1.1降至1.02,对应的反应收率从97.6%降至90.9%;而继续将POCl3当量从1.1提升至1.3对收率几乎无影响。说明POCl3适当过量有利于促进反应物酚(1)的完全转化。兼顾反应原料的成本,POCl3当量采用1.1为宜。至此,相比于间歇工艺,连续流操作不但提升了反应收率,时空产率(STY)更是提升了至少40倍。二者的对比如表1所示表1基于Coflore ACR反应器在处理沉淀反应时的优异表现,可以推测相比于超声波这种“高频低幅”的振动方式,“低频高幅”才更适合解决含固多相物系的稳定流动问题。对Coflore ACR反应器中物料的流动方式进行分析,将其抽象成如图10所示的10级CSTR串联模型,结合多级全混流模型和“闭—闭系统”所对应的轴向扩散模型,计算得到无量纲Pe数约为18.94,表明Coflore ACR反应器中的流型已经偏离了全混流,但仍远离平推流。图10分别在间歇式、浸没于超声水浴中的PFA盘管式以及Coflore ACR三种反应器中进行该反应并得到了相应的沉淀产物,利用扫描电镜(SEM)和激光粒度仪分别对颗粒的形貌和粒径分布进行表征,结果如图11、12以及表2所示。可以看出,颗粒尺寸基本都处于微米级。在间歇反应器中,沉淀颗粒偏向自由生长并形成球状外观,粒径分布总体上偏大。在另外两种连续流反应器中,粒径尺寸相近且总体偏小,只是Coflore ACR中的粒径分布更广,颗粒形状更无序和混乱;而PFA盘管中的颗粒形状更有序且总体偏向柱形。这意味着虽然Coflore ACR强烈的搅拌剪切作用也和超声一样可以减小颗粒的粒径,但最主要的作用还是避免固相颗粒在反应器壁上的黏附和沉积,从而保证颗粒均匀分散及稳定的两相流动。图11图12表2随着此反应在实验室规模反应器Coflore ACR中成功运行,工艺放大问题随之而来。若选用反应体积更大且依然能保持良好传质传热性能以及对多相反应具有出色耐受性的反应器,则连续多级搅拌管式反应器Coflore ATR是一个极好的选择。更难得的是,由于Coflore ATR流道的特殊构型,使其内部流动更接近平推流,返混更小,从而使反应效率更高。Coflore ATR反应器内最多可以串联8根反应管,每根1.25 L,反应体积共10 L,因此可将工艺放大至比Coflore ACR大100倍左右的规模。目前来看,这项工作是极有意义且充满希望的!【实验结论】本文作者第一次用连续流的方式合成了2,2’-亚甲基-双(4,6-二-叔丁基苯氧基)磷酰氯这一重要的聚合物成核剂前体,在此过程中使用基于多级CSTR原理设计的连续多级搅拌反应器Coflore ACR成功解决了反应沉淀堵塞流道的棘手问题,并将反应收率优化至高达98%,时空产率比间歇式反应提高了40倍以上。而连续搅拌技术也为类似问题提供了有效可行的解决方案。[原文]:https://doi.org/10.1021/acs.oprd.1c00105【公司简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20210830/20210830111814_9186.pdf
应用实例
2021.12.01

【南京工业大学案例】连续流光诱导可逆加成断裂链转移聚合反应和光介导聚合诱导自组装
【概况】微流体技术和光诱导可逆加成断裂链转移聚合(photo-RAFT)及光介导聚合诱导自组装(light-PISA)技术的结合,可以实现传统间歇反应器不具备的独特优势。最近基于微反应器photo-RAFT和light-PISA技术,许多具有精确化学结构的聚合物及具有高阶形貌的先进纳米材料被相继报道。南京工业大学郭凯课题组2020年Chemical Engineering Journal发表Review《Continuous flow photo-RAFT and light-PISA》。本文主要综述了连续流光诱导聚合、光引发RAFT聚合和光诱导电子/能量转移RAFT聚合反应(PET-RAFT)的研究进展,文中光化学反应有采用荷兰Chemtrix微通道反应器 Labtrix Start进行相关验证实验。此外,介绍了微流体中的light-PISA,并指出了目前所面临的挑战。我们希望能在流体化学、聚合物精密合成和纳米科学方面提供有用见解。【前言】2020年是聚合物科学100周年的里程碑。Hermann Staudinger于1920年发表的论文über Polymerisation及其后续相关研究成果被认为是“大分子假说”的基础。回顾历史发展,聚合物工业(1870年的胶片,1900年的酚醛树脂)推动了聚合物科学理论的发展,并反过来被这些理论所推动。目前,通过自由基聚合、配位聚合、离子聚合、开环聚合、缩合聚合等方法制备的聚合物已被广泛应用。当今世界的聚合物材料大多依据自由基聚合(FRP)反应。从化学工程的角度来看,对动量/热量/质量传递及反应过程的深入研究使得自由基聚合成为聚合物工业中最重要的方法。但是高浓度的自由基在使反应速度加快的同时,也会导致其分子量的分散。所以20世纪90年代以来,基于自由基的可逆失活原理,又出现了可控自由基聚合(CRP),也叫可逆失活自由基聚合(RDRP)。它包括原子转移自由基聚合(ATRP)、可逆加成裂解链转移聚合(RAFT)、氮氧化物介导聚合(NMP)等。图1 三种Photo-RAFT聚合的机理得益于Rizzardo和Moad等人于1998年开发的高效链转移剂(CTA),RAFT技术可以实现对聚合物化学结构和拓扑结构的有效控制,并且具有“条件温和、单体可选范围广及与其他反应相容性强”的优点。而且基于RAFT的聚合诱导自组装技术(PISA技术)也已经成功用于合成多种常规聚合物及不同形态的高固体浓度新型纳米材料。首先引发剂热激活产生自由基;然后光作为外部刺激,在室温下触发RAFT和PISA反应。根据机理的不同,photo-RAFT聚合反应可分为三种:(1)光引发-转移-终止聚合(无外源引发剂或催化剂);(2)带有光引发剂的RAFT聚合;(3)光诱导电子/能量转移RAFT聚合(PET-RAFT) (图1)。与热激活方式相比,photo-RAFT和Light-PISA在合成聚合物和纳米材料时具有独特的空间/时间控制优势(尤其是存在高温敏感的官能团时)。但进一步的学术研究和工业应用结果显示,光在反应器中的衰减问题对photo-RAFT和Light-PISA也构成了不小的挑战。与传统间歇反应器相比,微反应器内部尺寸小(最近,许多连续流photo-RAFT和Light-PISA的相关研究工作成功制备了多种具有精确微结构的聚合物和具有高阶形貌的先进纳米物。而且可以很容易地实现放大。本文对流体聚合、RAFT和PISA反应进行了综述。重点介绍了基于微反应器的光引发-转移-终止聚合、光引发剂的RAFT聚合以及PET-RAFT聚合。然后介绍了连续流中出现的Light-PISA反应。提出了该课题未来发展面临的机遇和挑战。我们希望能在流体化学、聚合物精密合成和纳米材料制备等方面提供有用见解。【连续流光引发-转移-终止聚合】表1 连续流Photo-RAFT聚合总结Photo-RAFT技术已成为聚合物合成的有效合成方法。表1汇总了典型的链转移剂、引发剂、光催化剂,及相关文献报道的单体、反应条件(光源和反应时间)、产物分子量和分散性结果。相关化学结构如图2所示。图2 单体、CTA、光引发剂和光催化剂的结构光引发-转移-终止聚合反应的一大特点是:无需外源引发剂和催化剂。通常在紫外线的照射下,CTA发生光裂解,产生碳中心自由基,从而引发单体的聚合(图1)。CTA既作为链转移剂,也作为 链终止剂参与聚合反应。随着反应的发生,CTA逐渐降解,这会导致聚合控制不良,尤其是在高转化率时。最后利用可见光介导聚合反应,有效控制分子量的分散度。Johnson等人以二苄基2,2’-(硫代羰基双(磺酰胺基))双(2-甲基丙酸)为CTA,利用连续流反应器提高了N-异丙基丙烯酰胺(NIPAM)、丙烯酸叔丁酯(tBA)、乙二醇甲醚丙烯酸酯(EGMEA)和N, N-二甲基丙烯酰胺 (DMA)之间的UV介导聚合反应的速率(图3A)。他们设计了装置A,将Halar管(内径为0.762 mm)包裹在紫外灯(Amax = 352 nm)上(图3B)。UV灯的开/关可以控制聚合继续进行和停止(图3D)。与间歇反应相比,连续流聚合速率增加了四倍(转化率74%,240分钟→转化率80%,60分钟)。但辐射时间较长时(>30 min),聚合度(DP = 500)提高,分散度也增加。这是由于UV辐射造成了不可逆的BTMP分解。为此,他们增加了紫外灯到反应器的距离,以降低光的强度。通过装置B(图3C),NIPAM在30分钟内的转化率为50%,得到了分布较窄的PNIPAM (M= 26.9 kg/mol, D=1.10)产物。通过调节辐射时间(即停留时间),分子量线性增大至41.6 kg/mol(图3F),同时分散度低于1.2(图3E)。而且在流动条件下, 400 min可得到2.95 g PDMA (M= 24.9 kg/mol, D=1.11)。这表明:通过延长停留时间可实现过程放大。图3 (A)以BTMP为CTA,在乙腈(MeCN)中紫外光照射下进行光引发-转移-终止聚合。(B) photo-CRP的连续流动设置A。 (C) photo-CRP的连续流量设置B。(D)流动条件下photo-CRP的“开”/“关”实验。(E) ln([M]0/[M]t)随辐射时间(流动中的停留时间)的变化,其中[M]0和[M]t分别是时间点0和t处单体的浓度。(F) Mn及Mw/Mn与单体转化率之间的关系。基于异戊二烯和苯乙烯的聚合物,如聚(苯乙烯-二戊二烯-b-苯乙烯)(SIS),是一类具有广泛应用的弹性体材料。工业上,这类聚合物主要通过阴离子聚合或乳液自由基聚合来生产。考虑到其热激活机理,Junkers等人在微反应器中进行了超快高温紫外线介导的异戊二烯和苯乙烯共聚反应(图4)。在120-145 ℃的温度下,通过控制紫外光的照射强度(Amax = 365 nm),调节异戊二烯的传递速率。与相同温度下的常规聚合相比,连续流聚合将反应时间从几天缩短到了几分钟(转化率78%,100分钟,145℃)。当微反应器的停留体积从19.5 μL增加到2 mL时,可以达到每天100 g到100 kg的高产量制备。另外,该微反应器可以实现异戊二烯和苯乙烯的嵌段共聚,成功制得了二嵌段共聚物聚苯乙烯-b-聚异戊二烯(PS-b-PI, Mn= 17.3 kg/mol, D=1.42)和三嵌段共聚物PS-b-PI-b-PS (Mn = 25.4 kg/mol, D=1.57)。后来,Junkers等人又采用连续流反应器研究了甲基丙烯酸酯的可见光催化聚合反应。制作了一个气密性的全氟烷氧基管状微反应器(ID = 0.75 mm)。以LED蓝光为光源(max = 450 nm),减缓了三硫代碳酸盐(TTC)的分解。停留时间在60 min以内,甲基丙烯酸甲酯实现了完全转化。产物分子量大于10 kg/mol),且相对分散范围较窄(D-1.20-1.30)。作为链延伸和嵌段共聚物合成的关键,端基的保护度也较高。而且该连续流反应可在12 h内收集约25 g的二嵌段共聚物。图4 异戊二烯和苯乙烯在连续流反应器中的光引发-转移-终止聚合3 连续流光引发RAFT聚合与光引发-转移-终止聚合相比,RAFT聚合的区别在于起始自由基是光引发剂生成的,而不是来自CTA的降解。I型光引发剂通过单分子裂解反应生成自由基,而II型光引发剂通过与氢供体(胺或硫醇)的双分子反应生成自由基。Gardiner等人以偶氮双(环己烷腈)(Vazo88)、2-苄基-2-(二甲氨基)-1-[4-(4-吗啉基)苯基]-1-丁酮(Irgacure 369)和苯基双(2,4,6-三甲基苯甲酰)氧化膦(Irgacure 819,TPO)为光引发剂,CDPTA为CTA,利用Vapourtec光流反应器(ID = 1.3 mm,停留体积= 10 mL)在紫外线照射下进行(甲基)丙烯酸酯和丙烯酰胺的RAFT聚合。在330 ~ 380 nm(滤光片4)的波长范围内诱导DMA聚合,得到了相应聚合物产物(Mn= 10.0 ~ 17.6 kg/ mol, D=1.14 ~ 1.60)。随着波长范围的扩大,聚合速率提高,分散度也随之扩大。高负载光引发剂和CTA时([DMA]:[CTA]:[光引发剂]= 100:2:2),30℃下仅需5分钟即可完成聚合,而heat- RAFT工艺需要更多的时间和更高的温度(>100C)。Junkers等人采用另一系列的光引发剂,以2-(十二烷基硫代硫代丙酸)(DOPAT)为CTA,(图5A),利用玻璃芯片微反应器(荷兰Chemtrix,停留体积为19.5 uL)与紫外灯(max =365 nm) (图5B)进行丙烯酸正丁酯(nBA)的RAFT聚合过程。研究发现:最佳反应条件([安息香] : [DoPAT]= 0.25: 1, 60 C, 30 mW/cm2)下,20分钟内的单体转化率可达85%。而且由于光引发剂分解速率的不同,安息香、2,2-二甲氧基-2-苯乙酮(DMPA)、Irgacure 819和2-羟基-4’-(2-羟基乙氧基)-2-甲基丙苯酮(Irgacure 2959)表现出来不同的动力学曲线(图5C)。图5 (A) 以DoPAT为CTA,在乙酸正丁酯(BuAc)中紫外光照射下用光引发剂进行RAFT聚合合成PnBA。(B) 所用微流反应器(Chemtrix Labtrix Start)示意图。(C) 不同光引发剂对应的nBAPhoto-RAFT聚合动力学一级图。Cheng等人提出用光引发剂改进连续流RAFT聚合。比如,水溶性的2,2'-偶氮双[2-(2-咪唑啉-2-基)丙烷]二盐酸盐(AIBI)在可见光(max = 390-630 nm)照射下可同时作为光引发剂和氧清除剂,从而可以省去预脱氧的步骤(图6A)。他们以4-氰-4(乙基硫代硫代硫代)戊酸(CETP)为CTA,对比了聚乙二醇甲基醚甲基丙烯酸酯(PEGMA)聚合反应在连续流反应器和间歇反应器中的差别。连续流聚合反应中,分子量随单体转化率线性增加,分散度保持在1.3以下(图6D)。在配有紫色LED灯(Amax=390 nm, 4.92 mW/cm2)(图6B)的石英管微反应器(ID = 2 mm)中,随着流速的增加,聚合速度也加快。与小玻璃瓶中的反应常数(KaPP = 0.63 × 10-3/s)相比,连续流的表观聚合常数提高到1.01×10-3/s(图6C)。此外,反应时间从30 min(间歇反应器)减少到8 min(微反应器)(图6C)。这是由于连续流反应器中光可以均匀而有效的穿透流动相。最终得到了纯度较高的PPEGMA (Mn= 15.1kg/mol, D=1.11),单体转化率约为85.5%。图6 (A) 以CETP为CTA,AIBI为光引发剂,在水中进行无脱气蓝光辐射RAFT聚合合成PPEGMA。(B) 连续管式反应器中可见光控制RAFT聚合的示意图。(C) PEGMA在管式反应器和间歇反应器中可见光控制RAFT聚合的动力学一级图。(D) 管式反应器和间歇反应器中,PEGMA可见光控制RAFT聚合的数均分子量(Mn)及分散度(Mw/Mn)与转化率的关系【连续流PET-RAFT聚合】PET-RAFT聚合正朝着“制造绿色和精密聚合物”的方向发展。Boyer等人对PET-RAFT聚合的发展做出了开创性的贡献。与传统的光引发-转移-终止聚合和光引发RAFT聚合相比,PET-RAFT聚合只需要低能量的可见光。这样就可以实现更高水平的时空、波长和强度控制。光氧化还原催化剂在可见光照射下进入激发态,然后通过PET过程还原RAFT剂,形成自由基(图1)。基态分子氧(1o)通过三重态-三重态亲和(TTA)转变为单重态(o)的能力导致PET-RAFT聚合具有出色的耐氧性。它简化了聚合过程,为行业的大规模生产降低了成本。铱和钌配合物是最原始的光氧化还原催化剂。后来又有一系列的有机染料,如四苯基卟啉锌(ZnTPP),被用于PET-RAFT聚合。最近,Boyer等人报道了一种pH响应的有机光催化剂,并将其用于pH和光的双门控聚合反应。丰富的催化剂可选性,超低的催化剂浓度和催化剂可回收性为PET-RAFT聚合带来了显著的绿色和可持续性。此外,PET-RAFT聚合还具有单体可选范围大、分子量小、分散性好、选择性强、端基保持度高、与其他合成条件相容性好等优点。2016年,Boyer等人以ZnTPP为光氧化还原催化剂将二甲基亚砜(DMSO)中耐氧PET-RAFT聚合从间歇反应器首次转移到连续流反应器中(图7)。通过TTA生成单重态氧并与DMSO反应,使聚合过程具有良好的耐氧性。聚四氟乙烯管状反应器(容积为8.7 mL, ID为1.5 mm)上安装了600个低能LED灯(Amax = 560 nm)。无需预先脱气,停留60 min,实现了92%的单体转化率,得到了聚(N, N-二乙基丙烯酰胺)(PDEAm)(最高为14.2kg/ mol, D图7 以DoPAT为CTA,ZnTPP为光催化剂,在DMSO中绿光照射下进行不脱气连续流动PET-RAFT聚合合成PDEAm后来,Boyer等人又在绿光(Amax = 530 nm)之下,以曙红Y (EY)和三乙醇胺(TEtOHA)为催化剂,开发了另一种连续流耐氧水性PET-RAFT聚合工艺。反应器装置由PFA管(ID = 0.75 mm)和PEEK混合器(外径(OD) = 1.59 mm)组成(图8C)。EY和TEtOHA在不脱氧的情况下,可在10分钟内实现95%的DMA转化,分散度为1.3。通过将目标DP从100增加到1000,可获得82.7 kg/mol的PDMA (D=1.81),停留时间为10 min,无抑制期(转化率为83%)。Mn与转化率之间呈线性关系,Mn与Mn,th之间有很好的相关性。这表明可以良好地控制反应过程。接着换用其他丙烯酸酯和丙烯酰胺,如2-羟乙基丙烯酸酯(HEA)和n-羟乙基丙烯酰胺(HEAA)。以DMA和2-n-(丁基三硫代碳酸酯)丙酸(BTPA)分别为单体和CTA进行链延伸(图8A)。在三个连续反应器(分别为0.5、1和3 mL)中(图8B),将流速调整为0.05、0.1和0.3 mL/min,PDMA100-b-PDMA50-b-PDMA50和PDMA10-b-PDMA10-b-PDMA10通过PET-RAFT水溶液聚合法制备PDMA10(图8D)。理论上,可在24 h内得到300 g三嵌段共聚物。图8 (A) 以BTPA为CTA,EY/TEtOHA为催化体系,在绿光照射下进行PET-RAFT聚合合成三嵌段共聚物。(B) 三个反应器直接耦合的流动反应器设置。(C) 带有注射泵的反应器1、带有注射泵的反应器2、微混合器三通和背压调节器(40 psi)的光流反应器装置。(D)水性RAFT光聚合合成三嵌段共聚物PDMA-b-PDMA-b-PDMA过程中,均聚物(DP10)和嵌段共聚物(DP20)扩链后的MWD演变。图9 合成半氟嵌段共聚物的连续流装置Chen等人报道了半氟化(甲基)丙烯酸酯在微反应器(ID = 0.762 mm)中连续流Photo-RAFT聚合工艺(图9)。以半氟三硫代碳酸酯(TTC-2F)为CTA,10-苯基吩噻嗪衍生物(PTH-2)为催化剂,暴露在白色LED灯(Amax = 400 - 750 nm)下30min之后,实现完全转化,得到了窄分散的聚(丙烯酸六氟丁酯)(PHFBA)(Mn=4.7kg/mol, D = 1.06)。丙烯酸甲酯(MA)在80%的转化率下进行扩链反应得到PHFBA-b-PMA (Mn=10.8 kg/mol, D=1.07)(图9)。还研究了混合对流动反应器中PET-RAFT聚合链延伸的影响(图10A)。如图10B所示,Y型混合器的混合效率较低,嵌段共聚物扩链后的分散度大致为1.30-1.89。而通过填料塞混合器混合,立即就会形成均匀的混合物(图10C)。对多种CTA和单体而言,填料塞混合器的分散性度均比Y型混合器要低(图10D)。当改变CTA和单体之间的流速比(1:2,1:1,5:1)时,填料塞混合器中的分子量(Mn = 14.6-15.2 kg/mol)和分散度(D-1.20-1.26)几乎不变,而Y型混合器则存在较为明显的偏差(图10E)。而且对每个流量比而言,填料塞混合器的分散度都小于Y型混合器的分散度(图10E)。这主要是得益于填料塞混合器的高混合效率。图10 (A) 通过Photo-CRP进行链延伸的连续流装置。(B) 使用Y型混合器时的混合行为。(C)使用SiO2填充混合器时的流动混合行为。(D) 用常规混合器或填充塞混合器时的macro-CTA1、macro-CTA2和macro-CTA3嵌段共聚物表征。(E) 不同流量比的扩链实验结果分子量分布(MWD)对高分子材料的性能有显著影响,所以MWD控制引起了越来越多的关注。Boyer等人报道了一种通过连续流PET-RAFT聚合控制MWD形状的方法。以DMSO中ZnTPP催化的以DoPAT为CTA的DMA聚合反应为目标研究模型。PDMA的分子量可以比较容易地通过调节光强、CTA浓度和低速率来控制。然后收集和混合这些单组分的聚合物,可形成特定的MWD。通过改变停留时间分布、光变化(Amax = 460,530, 630 nm)及流动中的化学物质浓度来调控MWD的形状。图11 全连续和柱塞流流型及相应产品管式微反应器中的层流分布(Re相对较大的雷诺数(1929.5 vs 1-4.1)可以使流体剖面接近过渡流,从而避免完全连续流动下的分散问题(图12B)。设计的MWD在目标分子量范围(15.0-35.6 kg/mol, 15.0-63.2 kg/mol)内具有相同的响应(图12C)。更重要的是,通过向第一嵌段引入第二单体溶液,可选择性地延长所得分离聚合物部分的链段,以产生具有组成梯度的嵌段共聚物(图12 d和E)。GPC上定制MWD向单峰峰值的移动证实:具有不同分子量的所有单个组分选择性扩链后获得了相似的分子量(图12F)。而且GPC上单峰峰型转变为扩宽型(图12F)的事实也进一步证明了MWD的可控性。总之成功连续制备了具有可控MWD的聚(N,N-二甲基丙烯酰胺)-b-聚(N-丙烯酰吗啉)(PDMA-b-PNAM)和聚(N,N-二甲基丙烯酰胺)-b-聚(丙烯酸苄酯) PDMA-b-PBzA共聚物。图12 (A) 以BTPA为CTA,ZnTPP为光催化剂,在DMSO中绿光照射下PET-RAFT聚合DMA;(B) PDMA的定制MWD;(C) 由四种不同分子量的聚合物组成的定制MWD 1及由六种不同分子量的聚合物组成的定制MWD 2;(D) 通过PET-RAFT聚合将链延伸至PDMA-b-PDMA;(E) 通过单程流动过程定制产物成分和MWD;(F)最初广泛分布的缩小和最初狭小分布的扩大。聚合过程中会观察到粘度的增长,特别是对于高固相含量的聚合物。这不仅会造成流动反应器的堵塞,而且会造成较宽的停留时间分布。管中心的层流层分子量低(停留时间短),而反应器壁上的流体分子量高(停留时间长),这导致聚合物分散高。为此,Chen等人将液滴引入PFA管状微反应器(ID=1.0 mm),开发了一种滴流式PET-RAFT聚合工艺。在完全连续流动操作(40 L/min)下,随着ZnTPP催化DMA (IDMA]:[CTA]: [ZnTPP1 = 200: 1:01 in DMSO)聚合的固体浓度从18 wt%提高到48 wt%,分散度增加到1.30-1.68(图13A)。可见,滴流反应器可以在相同的停留时间(25分钟)内实现高固相浓度(48-78 wt%)、相似转化率(>80%)和窄分散(还使用了其他丙烯酸酯和酰基酰胺,如HEAA和2-羧乙基丙烯酸酯(CEA)。在不同浓度(48-78 wt%)和规模(3.4 g/60 min PDMA (Mn=17.5 kg/mol)之间实现了自动切换(Mn = 17.5 kg/mol, ?=1.21)。不同时间的一系列样品对每种浓度表现出完全相同的分子量和分散性(图13D)。利用这种自动滴流系统,可在高浓度固体(高达99%)条件下制备聚合物。图13 (A) 以4-氰基-4(己基硫氧甲基硫基)戊酸(CHP)为CTA,ZnTPP为光催化剂,在DMSO中绿光照射下PET-RAFT聚合DMA;(B) 聚合的流动反应器装置;(C) 流动反应器装置的光学照片;(D) 不同浓度和规模之间的自动切换。为了实现大规模且稳定的聚合物生产,Boyer等人研究了流动PET-RAFT聚合,并比较了两种流动模式(全连续流动和柱塞流动)的差异。在四种市售有机染料中,以红色素E (EB)为光催化剂,BTPA为CTA,在不脱气的情况下进行聚合反应(图14A)。反应器的停留体积为28.2 mL,停留时间为4 h(总运行时间为12 h)。当[DMA]:[IDMA]:[BTPA] =199.4:1,50 ppm EB, VDMA/VDMSO=30/70时,这两种流动模式表现出明显的差异。在完全连续流操作下,初期的分子量保持相对恒定。但当收集时间达到570分钟后,分子量急剧增加,分散度也变宽(图14B)。相比之下,柱塞流操作的产物分子量(Mn= 21.9±0.3 kg/mol)和分散度(D=1.09-1.12)从起始到结束几乎没有变化(图14C)。此外,在相同的停留时间下,柱塞流比完全连续模式(Mn= 19.4 kg/mol, D=1.41 vs Mn= 22.1 kg/mol, B-1.09)获得了更高、更一致的单体转化率(图14D)。这主要得益于段塞流的循环流特性及透光均匀性。而层流则导致了停留时间分布和透光梯度,特别是对于高粘度的反应混合物。考虑到柱塞流的优势,该小组又开发了一种高通量合成具有抗菌性能的共聚物的工艺。在初步的小试研究后探索了构效关系,最后实现了柱塞流PET-RAFT聚合的大规模生产(27.2 g/d)。图14 (A) 以BTPA为CTA,EB为光催化剂,在二甲基亚砜中进行PET-RAFT聚合,在绿光照射下合成PDMA;(B) 全连续过程中Mn和?随时间的变化以及不同时间采样点处的GPC结果;(C) 柱塞流过程中Mn和?随时间的变化以及不同时间采样点处的GPC结果;(D) 全连续和柱塞流过程的瞬时单体转化率及过程累积MWD的GPC结果。【连续流Light-PISA聚合】两亲性嵌段共聚物在溶液中的自组装可以形成各种纳米材料。传统的选择性溶剂透析方法耗时长(几天),且固体含量低(PISA聚合通常在几分钟到几小时内完成,固体含量也可高达50%。理论上,所有可控聚合都可以应用PISA工艺。其中,RAFT聚合由于单体范围广、条件温和,是目前应用最广泛的PISA方法。最初,大多数案例都是间歇反应器和微反应器中的thermal-RAFT PISA反应。而最近,photo-RAFT和微流体技术被引入开发连续流Light-PISA工艺。图15 (A) 以mPEG-CDTPA为CTA,在绿光照射下在水中进行光引发聚合合成PDMA-b-PHPMA;(B) 用于合成PEG-B-PHPMA纳米颗粒的能够监测颗粒形态的反应器装置;(C) 以BTPA为CTA,EY/TEtOHA为催化体系,在绿光照射下PET-RAFT聚合一锅法合成PDMA-b-(PDAAM-co-PDMA);(D) 耦合两个反应器的流动反应器装置示意图及PDMA-b-(PDAAM-stat-PDMA)纳米颗粒的TEM照片,该合成以17.5 wt%固体含量延伸PDMAA和DP200、DP400和DP600(BW=分支蠕虫,HBW=高度分支蠕虫,V=囊泡)。Zetterlund等人2018年开创了连续流Light-PISA技术。以聚乙二醇(PEG)封端的三硫代碳酸酯(mPEG-CDTPA)为CTA,甲基丙烯酸羟丙基酯(HPMA)在蓝光(Amax = 460 nm)下发生水性光引发-转移-终止聚合,形成PEG-b-PHPMA纳米颗粒(图15A)。光强(0.8 mW/cm2和1.6 mW/cm2)、固含量(15-20 wt %)和停留时间(15-90 min)对PISA的影响研究结果表明:强光强可以加快聚合速度;高固含量可以使1mL PFA管式反应器(ID=1 mm)中出现纯囊泡状纳米颗粒。在最佳工艺条件(1.6 mW/cm2, 20 wt%固含量)下,微反应器停留75 min,间歇反应器停留6 h,可获得单体完全转化的PEG-b-PHPMA (Mn=58.0 kg/mol, D=1.62)。通过增加HPMA的DP,观察到了蠕虫状到囊泡状的形态演变 (50 - 200 nm)。有趣的是,连续流动过程中获得的空心斑片状囊泡(DP = 400),在相同条件额间歇式反应器中无法获得(图15B)。这是由于流动状态下的反应条件更加精确。通过连续流动PET-RAFT PISA合成PDMA-b-(PDAAM-co-PDMA)纳米颗粒时也观察到了这一现象。虽然通过增加流速没有显著的形态变化,但使用微流体技术的一大优势是可以通过简单地增加停留体积来监测形态演变(图15B),并且可以通过延长周期来扩大产量。后来,他们又通过尝试了更多的方法来扩展了连续流photo-RAFT反应,如PET-RAFT聚合工艺。以曙红Y/三乙基乙醇胺为催化剂,在绿光(max = 530 nm)多级流反应器中,不分离CTA,连续进行DMA的PET-RAFT水溶液聚合及随后的双丙酮丙烯酰胺(DAAM)/DMA的链延伸(图15C和D)。加快了聚合速度,形成了片状囊泡和放大合成,并有效地制备了基于聚丙烯酰胺的纳米颗粒。他们还将研究范围从水系扩展到了醇系。低极性醇(乙醇/共溶剂)介质中,蓝光(Amax = 460 nm)条件下,通过光致发光技术制备了不同形貌的聚(聚乙二醇)甲基丙烯酸甲酯-b-聚甲基丙烯酸苄酯(PPEGMA-b-PBzMA)纳米粒子。意外的是,加入一系列光引发剂或者增加光强,都未能提高聚合速率。除TPO光引发剂外,只有使用丙烯酸酯基的大分子CTA和丙烯酸酯单体才能实现快速聚合。PISA技术的难点之一是对氧气的高灵敏性。所用PISA过程通常需要脱气,且在惰性气氛中进行。在这方面,Tan等人开发了一种可以在水和有机介质及广泛的温度范围内进行的耐氧Light-PISA工艺。其原理为双波长光诱导脱氧和I型光引发。以曙红Y和ZnTPP为光氧化还原催化剂,以苯基-2,4,6三甲基苯甲酰膦酸钠(SPTP)和TPO分别为水和醇/水环境中的I型光引发剂。他们设计了管式反应器系统,其中绿色LED灯包裹在两个结晶盘的外层,紫色LED包裹在第二个结晶盘的内层(图16D)。对以mPEG-CETP为CTA的HPMA水溶液RAFT聚合反应而言,[IHPMA]:[mPEG-CETP]:[Eosin Y]:[AscA]:[SPTP]= 300:1:0.05:1:1/3)时(图16A),单次紫光照射(Amax =405 nm, 1.0 mW/cm2)半小时后单体转化率仅为36% (图16B)。这是由于氧的抑制作用。相反,在绿光照射15分钟后,在紫光照射15分钟内就获得了差不多的单体转化率(图16B)。均相和非均相两阶段半对数图显示,在保持分子量低分散性的情况下,实际分子量与理论分子量一致(图16C)。随着停留时间的增加,分子量随转化率线性增长(图16E),GPC轨迹向更高的分子量移动(图16F)。结果表明,曙红Y/抗坏血酸(AscA) (Amax = 530 nm)光诱导脱氧是有效的,可使后续与SPTP光引发剂(Amax-405 nm)的RAFT聚合速率加快。这一方法也被证明适用于醇类系统。在两个反应器中,通过调节固含量和PHPMA的DP,观察到了不同的形貌(图16G和H)。而且,在间歇式反应器中很容易形成更高阶的形貌(图16H)。例如,在流动系统中只得到球状,而固体含量为15%、DP为198的釜式反应器可以得到囊泡和蠕虫状。反应器类型对嵌段共聚物的组成没有影响。微反应器内的流体动力学和间歇式反应器内的搅拌区别是造成上述形貌差异的主要原因。图16 (A) 以mPEG-CETP为CTA,EY/AscA为催化体系,SPTP为I型光引发剂,紫光和蓝光辐射下PET-RAFT聚合合成PEG-b-PHPMA;(B) 通过双/单波长光辐射([HPMA]:[mPEG-CETP]:[Eosin Y]:[AscA]:[SPTP]=300:1:0.05:1:1/3)在连续流反应器中进行的HPMA(15%w/w)水性光PISA的聚合动力学;(C)在连续流反应器中进行的HPMA(15%w/w)的双波长水性光PISA图;(D) 用于双波长Light-PISA的流动反应器的照片和示意图;(E) 水性双波长光PISA中Mn和Mw/Mn随单体转化率的变化;(F) HPMA(15%w)双波长水性光PISA形成的二嵌段共聚物的GPC结果;(G) 连续流反应器中双波长水性光PISA制备的mPEG-PHPMA二嵌段共聚物纳米物体的TEM照片;(H) 间歇反应器中用双波长水性光PISA制备的mPEG-PHPMA二嵌段共聚物纳米物体的TEM照片。【总结和展望】目前连续流Photo-RAFT和Light-PISA技术的相关研究主要集中在将微反应器与光化学组合起来。与间歇反应器相比,连续流反应器中的Photo-RAFT和Light-PISA反应表现出诸多优点,如:更快的表观聚合速率、更容易扩大规模和更多的高阶形貌。在这些成果的鼓舞下,我们将在基础研究和工程研究方面付出更多努力,以推动连续流Photo-RAFT和Light-PISA反应的工业化。从化学工程的角度看,流动动力学参数是非常理想的。在微流动条件下,Photo-RAFT反应的速率系数、扩散限制和链长依赖关系等需要通过实验和模拟共同来研究。流动模式的数学模型是描述聚合和自组装过程的必需环节。在人工智能(AI)的协助下,我们期望开发出更好的建模策略。微反应器的放大效应在聚合动力学领域也是一个重要的研究课题。Guo等人发现了一个有趣的现象,即:随着光诱导有机催化ATRP微反应器内径的减小,表观聚合速率先增加后略有下降。这表明:并不是所有的微反应器的特征尺寸越小,性能越好。与完全连续流相比,柱塞流表现出有趣的结果,值得更密切的关注。为了开发新型功能聚合物和纳米物,更多类型的Photo-RAFT和Light-PISA工艺应该被研究。葡萄糖氧化酶(GOx)或吡喃糖2-氧化酶(P20x)作为光酶催化剂、氟化交替共聚物的光合作用和光控HAT-RAFT被认为是未来微反应器的研究目标。混合单体原料的选择性共聚是制备嵌段和多嵌段共聚物的长期挑战。而通过在连续流中耦合Photo-RAFT、正交聚合和外部刺激,有望实现这一目标。微反应器中的Light-PISA被证明可以获得更均匀的纳米结构和更新颖的形貌。形态的演变对于深入理解PISA及其潜在应用(如药物输送)具有重要意义。流动条件可能有助于捕获其中间形态及其演化情况。我们相信连续流Photo-RAFT和Light-PISA领域将出现越来越多有趣的研究发现。参考文献:[1] Continuous flow photo-RAFT and light-PISA[J]. Chemical Engineering Journal, 2020:127663.【公司简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20210906/20210906141448_0012.pdf
应用实例
2021.11.29

【Graz大学CCFLOW案例】使用比利时Creaflow振荡连续流反应器在水胶束条件下进行的连续流动非均相催化还原胺化反应
【背景介绍】由于无溶剂反应在合成化学中应用较少,通常仅限于少数特殊情况[1],而有机化学合成中产生的废物又以溶剂为主要成分[2]。因此,以水作为溶剂引起了人们对更可持续的生产实践的极大兴趣[3]。与传统有机溶剂相比,水作为天然丰富的反应介质在环境、安全和经济方面具有明显的优势[4]。然而,由于大多数有机化合物的不溶性和许多反应对水分的敏感性,水通常不被作为溶剂采用。为了克服这些问题,人们广泛研究了水胶束条件,即使用各种表面活性剂形成胶束阵列作为两亲性纳米反应通道[5]。这种胶束的亲油部分可作为“有机溶剂”发挥作用,建立非水相环境。通过这种方式,水敏感反应甚至也可以在水介质中进行[6]。据证实,胶束的粒径和形状在这种转变中至关重要[7],因此人们设计了新的表面活性剂以促进胶束性质的改善,并在水胶束条件下实现了一系列化学反应,包括各种交叉偶联、C-H活化、酰胺键的形成、氧化和还原等。在胶束化学中,表面活性剂分子的聚集体可形成胶体悬浮液,产生两相混合物,其中反应物的质量转移是有限的[3a]。如果胶束化学反应是在多相催化剂的存在下进行的,这种情况尤其明显,因为这些催化剂必须与纳米胶束疏水囊中的基质保持紧密接触[9]。水胶束催化通常是在传统的间歇釜式条件下进行的,其中混合和传质效果取决于反应釜的尺寸大小[5,8]。因此,由于这些限制,生产放大是一个很大的挑战[10]。相比之下,连续流反应器提供了独特的优势,可以显著改善多相之间的传输效果,并由于其特殊的表面积/体积比,传热效果也大大增加[11]。因此,过程强化以及生产的放大化都变得简单可行,同时,还可以通过控制反应参数来更好地提高产品质量和稳定性。然而,由于众所周知的堵塞和反应器污染问题,在流动反应器中处理固体物料(如泥浆和悬浮液)被视为连续处理中的主要挑战之一[12]。尽管目前研究人员已经开发了许多反应器来处理固液两相混合物[13],但最近只有一个在流动条件下进行多相胶束催化的例子,其中Suzuki–Miyaura耦合是在Fe/Pd纳米颗粒作为非均相催化剂的情况下利用一系列连续搅拌釜式反应器(CSTR)实现的[14]。尽管CSTR提供了在流动条件下处理固体物料的实用解决方案[15],但放大每个单元的反应釜尺寸可能会导致较差的混合效果。因此,作者的目的是采取一种不同的方法,开发一种使用振荡塞流反应器(OFR)在水中进行多相胶束催化的稳定可放大的连续过程。根据作者先前在处理OFR中含固体的连续流光催化过程的经验[16],作者推测,通过使用叠加在反应流净流量上的脉动产生的剧烈混合将会使传质效果增强,同时可以确保反应器内部水相中固液混合物实现稳定悬浮。此外,通过优化脉动参数,作者建立了一个稳定使用的流程来处理两相胶束介质,以实现最小的轴向分散和提高停留时间分布结果。奥地利格拉茨大学连续流合成与研究中心(CCFLOW)的Wernik等人描述了使用振荡连续流反应器(HANU-OFR)在水相胶束介质中进行非均相催化还原胺化连续流反应。该连续流工艺使包括氨基酸衍生物在内的多种底物能够在相当温和的条件下成功转化,增强了反应过程的混合传质效果,有效地改善了连续流工艺的高效、稳定和生产的放大性。此外,文章还证明了连续流工艺的生产能力以及多相催化剂和表面活性剂水溶液的可持续性。本文选择了还原胺化作为流程验证的模型[17],这是C–N键构建的关键方法之一,在药物和药物化学中起着至关重要的作用[18]。在初始亚胺形成过程中产生了化学计量的水,因此在水中进行的还原胺化在一开始是没有效果的[19],Lipshutz及其同事最近报道了一种在水胶束条件下进行还原胺化的间歇釜式反应过程[20]。在这项研究中,Pd/C作为一种现成的多相催化剂,Et3SiH作为还原剂,自行设计的表面活性剂TPGS-750-M,包含亲脂性维生素E部分与亲水性聚乙二醇(PEG)侧链结合(图1A)[21]。作者以这些文献研究结果作为研究的起点,开发了一种新的连续流动方案,用于实现胶束化学的水中非均相催化还原胺化反应(图1B)。图1. 表面活性剂TPGS-750-M(A),和连续流反应基本方案【实验结果与讨论】首先,作者使用苯甲醛与苯胺的还原胺化作为简单模型,在1.2 equiv.的Et3SiH和不同量的5 wt% 的Pd/C催化剂的存在下,对最重要的反应条件进行釜式筛选实验(表1)。尽管直接使用氢气可能更具原子经济性,但作者选择Et3SiH作为还原剂,主要是考虑到安全性和生产的可放大性,而直接使用气体试剂会使混合和传质在非均相胶束条件下更具难度。当研究不同两亲物质的影响时,相应的亚胺中间体在所有反应中都很容易得到,但随后被还原为1a,这证明了其高度依赖于所采用的表面活性剂(条目1-5)。实验表明,在50°C下反应60分钟后,TPGS-750-M的效果优于其他表面活性剂以及水相(无表面活性剂)反应。事实上,SDS、Brij S 100和Kolliphore EL并未产生任何显著的胶束加速效应,其结果与水相反应(转化率90–97%和选择性17–32%)相当,这可能是由于表面活性剂的不良亲水-亲油平衡和/或形成的胶束尺寸不足所导致的。令人满意的是,500 ppm的催化剂负载量证明是足够的(条目5-8),这与早期的金属催化还原胺化反应相比显著降低,甚至低于已报道的胶束条件下还原胺化反应的量[17,20]。催化剂极低负载量可能是因为TPGS-750-M形成了含有PEG的纳米胶束,把周围的钯纳米颗粒聚集起来,从而促进催化剂和反应物在胶束内部的紧密接触[9]。当TPGS-750-M的质量分数为2wt%时,反应效果最好(条目8);仅使用1 wt%的表面活性剂会导致形成胺显著减少(条目9)。成功还原亚胺中间体需要温和的加热条件,因此50 ℃或60 ℃被视为最佳值(条目8、10和11)。除活性炭外,作者还对各种催化剂载体进行了釜式筛选试验。SiO2、BaCO3和Al2O3显示出了与活性炭类似的结果,而BaSO4显示出了亚胺的不完全还原结果。表1. 釜式实验筛选重要反应条件的结果a 1mL 容积,转速500 rpm。b反应原料转化结果,基于GC-FID面积 。c选择性,基于GC-FID面积。(检测出亚胺是唯一的副产物) d 1wt% TPGS-750-M。e30分钟反应时间.在充分了解不同反应条件对苯甲醛和苯胺在水胶束条件下进行的釜式还原胺化的影响后,接下来在1 mL到100 mL的不同反应体积下进行该反应,目的是研究混合效应以及在放大过程中可能出现的问题。如图2所示,尽管在放大反应时调整了搅拌速度,但产率仍随着反应容积的增加而急剧下降。例如,在800 rpm的搅拌速度下,反应在1 mL容积时产生了92%的1a;然而,当体积分别增加到10ml、50ml和100ml时,产率急剧下降至62%、18%和14%。另外,在相同规模的反应中,产率明显随搅拌速度的增加而增加。这些结果表明,两相胶束反应受混合效果的影响十分显著,这意味着在釜式条件下进行生产放大存在明显的局限性。事实上,在大规模应用胶束反应技术的典型实例中,有机混合溶剂的使用可以用来改善釜式反应介质的物理特性,从而促进过程的传质效果。图2. 不同反应容量条件下苯甲醛和苯胺在水胶束中的釜式还原胺化反应研究(产率由GC-FID 面积百分比计算所得)在釜式反应数据的基础上,作者接下来进行连续流动方案的研究。为此,作者采用了连续流设备OFR(HANU? HX 15连续流反应器(Creaflow),该反应器包括一个线性板状反应通道,配有一系列立方静态混合元件。除了一个反应组分净流量的计量泵外,该系统还包含一个振荡隔膜泵,用来提供可调节的对称脉冲。通过与静态混合元件配合,可以实现反应流体的不断分离和重新混合。此外,振动电机有助于颗粒稳定地悬浮在体系通道中,而狭窄的反应通道则增加了脉动的混合速度,从而降低了即使在低流速下颗粒沉降的风险。在实验案例中,反应混合物的料浆通过蠕动泵连续泵入反应器中。该体系还包括一个3 bar的背压调节(BPR),用来防止在反向脉动期间出现气泡或从反应器出口吸入空气。这种HANU反应器最初设计用于多相流光化学反应[16,23]。本文表明,通过正向和反向混合的有效配合,连续流反应器中的传质效果与间歇过程和CSTR相比得到了显著改善。该反应器可以确保双相反应介质沿着反应器实现稳定悬浮,从而为在连续流动条件下进行水胶束反应提供一个稳定且可放大的操作工艺。其流程设计如图3所示。图3. 水胶束条件下连续流动胺化还原反应流程设计配置好OFR连续流反应器后,作者进行了连续流优化实验,主要研究振幅(每泵冲程的排量,mL)和频率(每秒冲程数,Hz)的影响,这也是该反应器独有的工艺参数设置。这些参数的调节可以使作者获得两相反应混合物的稳定悬浮液,同时实现反应区的轴向的最小化分散。对于OFR中的停留时间分布,作者根据文献结果进行了控制实验,通常较低的振幅意味着较窄的分布曲线,而频率也具有类似的趋势,但程度较低。在研究脉动参数对在TPGS-750-M作为表面活性剂存在下,Et3SiH介导的、Pd/C催化的苯甲醛与苯胺的还原胺化反应的影响后,实验发现,在较低振幅下,转化率不稳定,且形成强烈的泡沫(表2,条目1和条目2)。不过,如果将振幅增加到0.24 mL(约为置换体积的50%),则可以实现定量和较好的选择性反应,同时也能够保持反应混合物在反应器中的稳定悬浮(条目3)。为了进一步减少泡沫的形成,频率被进一步降低至0.6 Hz(条目4)。在非均相催化胶束条件下,连续流工艺发展的主要挑战之一是颗粒在反应器中的不定时沉降和沉积。文章认为,除了设置脉动参数外,反应悬浮液的稳定性还可以通过改变反应中所使用的固体的性质来进行改善。因此,在连续流动条件下作者重新评估了釜式实验中显示出良好效果的催化剂载体的影响。尽管使用5 wt%的Pd/Al2O3和5 wt% 的Pd/BaCO3(两者均为500 ppm)在流动条件下生成的胺1a具有定量的转化率和很好的选择性(分别为62%和95%),但由于这些载体材料的高密度(Al2O3: 4.00 g mL-1;碳酸钡:4.49g mL-1),在这两种情况下,颗粒沉降和堵塞的问题都会严重影响实际应用。相反,使用5 wt%的Pd/C(活性炭密度:2.01 g mL?1)时,反应运行稳定且无堵塞的发生,同时定量选择性地生成了产物1a(条目7)。在比较1 wt%和5 wt%的Pd/C(两种情况下均为500 ppm Pd)后,悬浮液的稳定性未发现存在明显的差异,因此,作者使用5 wt%的Pd/C进行了进一步的试验(条目8)。在进一步优化反应条件时,采用了250 ppm 5 wt%的Pd/C并将停留时间减少到20 min,这导致产率有所降低(条目9和条目10),因此最佳条件被确定为500 ppm的催化剂负载和30 min停留时间(对应于500μL min?1流量)。对于在流动模式下除TPGS-750-M以外的其他表面活性剂的影响,实验观察到了与釜式实验中类似的下降趋势,以及偶尔堵塞现象的存在。为了证明该工艺的适用性,接下来作者在优化后的胶束连续流条件下将一系列的醛和胺进行还原胺化[24]。除了苯甲醛,其4-氟代和4-甲氧基取代衍生物在还原胺化反应中均与苯胺发生了定量的选择性转化,以较高的收率(分别为84%和81%;图4A)生成了相应的仲胺(1b和1c)。1-萘醛在与苯胺的反应中表现出较低的反应活性;然而,当催化剂负载量增加到1000 ppm时,胺1d可以成功得到58%的分离收率。糠醛和2-苯丙醛表现出了较高的转化率和高选择性,生成胺1e和1f的产率分别为73%和61%。各种脂肪族醛与苯胺的还原胺化反应也显示出了良好的结果。2-乙基己醛反应平稳,以75%的产率生成胺1g。在环戊烷和环丙烷碳醛的反应中,相应的胺(1h和1i)分离收率均在70%以上,然而,在这些情况下,催化剂负载量也相应增加到了2000 ppm。本文使用苯甲醛作为反应基体研究了胺的不同种类(图4B)。其中,各类取代苯胺衍生物以及苄胺和苯乙胺具有优异的转化率和高选择性,并以70–89%的产率生成了合适的胺(2a–d)。各类脂肪族胺,包括开链、α-和β-支链以及脂环伯胺的还原胺化反应在流动条件下都能够顺利进行。这些反应的选择性均不小于90%,并且相应的仲胺产物(2e–h)的分离收率高达85%。此外,仲胺吡咯烷被定量选择性地转化为叔胺2i,产率79%。值得注意的是,对于所研究的大多数胺底物,催化剂负载量相应地增加到了1000或2000 ppm。表2. 连续流反应条件优化实验a25 mL 反应体积。b脉动振幅(每泵冲程的排量,mL)。c频率(每秒冲程数,Hz)。d反应物的转化率,按GC-FID面积%计算所得。e选择性,GC-FID面积%计算所得(相应的亚胺被检测确定为唯一的副产物)。f250 ppm催化剂。g稳定性实验,60 mL反应体积。h流速为500μL min-1。i流速为750μL min-1。图4. 水胶束中Pb/C催化连续流动还原胺化反应底物类型研究(转化率和选择性由GC-FID面积%计算所得;产率为分离收率)a1000 ppm 5 wt% Pd/C。b2000 ppm 5 wt% Pd/C。 c温度 65℃天然氨基酸经常被用作各种生物活性物质和活性药物成分合成中的手性构建模块[25]。为了进一步验证本试验工艺的适用性,作者在胶束连续流条件下通过苯甲醛与L-苯丙氨酸的还原胺化反应制备了一系列的N-苄基化氨基酸衍生物,L-亮氨酸、L-脯氨酸和L-蛋氨酸甲酯(图4C)。结果表明,目标产物(3a-d)均可以在连续流反应器中成功制备,产率高达75%;然而,在这些情况下,亚胺的形成比简单结构底物的反应更加困难(图4A和B),例如产生了苯甲醇作为醛还原产生的副产物。在这些反应中,催化剂负载量通常设置为2000 ppm,在某些情况下,温度升高至65℃。为了验证所开发连续流工艺流程的稳定性和生产力,作者将4-氟苯甲醛与苯胺还原胺化试验进行了长时间的运行。考虑到起始原料溶液的粘度和密度可能会由于体系中亚胺的形成而随时间有所变化,试验使用了改进后的三股进料体系,其中,含有水性表面活性剂和Pd/C催化剂的悬浮液作为进料1,醛和还原剂的无溶剂混合物作为进料2,苯胺作为进料3(图5A)。所有其他反应条件均设置为之前优化后的参数。在稳态条件下监测7小时的反应,每小时检查一次转化率和选择性[26]。结果表明,实验期间证明该连续流体系非常稳定,没有发生堵塞或其他问题。所有样品均实现了定量转化和≥ 98 %的选择性,萃取后,简单的二氧化硅过滤足以去除任何多余杂质,从而获得分析纯产品。因此,最终产品的分离收率可以由所有样品的总量确定为15.0 g的胺1b(总收率85%)。这时的时空产量(STY)为143 g?1 L?1的,生产效率为2.14 g h?1。实验期间没有发生泄露损失,粗产品中测得的钯含量为1.4 ppm,硅胶过滤后,钯含量降低到了为了进一步提高当前连续流工艺的可持续性,作者尝试在长时间地运行相同的反应和条件时,回收利用多相催化剂和表面活性剂溶液。在每个循环之间,用乙酸乙酯萃取反应混合物中的胺产物。过滤催化剂,并用少量的EtOAc和水清洗,在室温下干燥,然后在下一次循环中再次使用。在重复使用表面活性剂溶液之前,需要在减压下从中去除残余的有机溶剂。通过这种方式,每次运行可回收91–95%的Pd/C以及>99%的表面活性剂溶液(见表S3?)。重要的是,在连续五次循环运行期间,反应仍可实现定量转化,且选择性稳定保持在>98%(图5B)。图5. 最优连续流条件下长时间运行试验(A),同一反应中催化剂和表面活性剂溶液的回收(B)【本文结论】1. 在非均相催化胶束条件下,本文用比利时Creaflow连续流反应器HX15研究了还原胺化连续流反应过程。该连续流工艺以Et3SiH为还原剂,Pd/C为多相催化剂,TPGS-750-M为市售表面活性剂。2. 由于振荡流模式和多个静态混合元件的有效结合,该连续流动体系被证明是在水胶束条件下进行反应的一个稳定且可放大的工艺,同时也可以有效增强不同反应相态之间的混合和传质效果。3. 通过优化重要的反应条件(如脉动振幅、频率、温度、停留时间和催化剂载体等),还原胺化反应在相对温和的条件下实现了高收率和高选择性,同时反应器中两相胶束介质的悬浮液一直保持在稳定状态。4. 一系列醛与各种胺的还原胺化反应已经在连续流反应器中得到实现,其中包括许多氨基酸衍生物。5. 该连续流程的生产实用性已通过一个7小时的长时间运行得到验证,最终实现了克级规模的生产能力和极低的废物排放量。同时,通过连续5次成功回收多相催化剂和表面活性剂水溶液,进一步阐述了该连续流工艺的可持续性。6. 这是在OFR连续体系中首次进行水胶束催化反应。值得注意的是,本文开发的连续流动工艺通过使用市场上中试规模的反应器(HANU)提供了一种简单直接的生产放大策略,该反应器保持了所有主要工艺特征,包括通道尺寸、停留时间分布和传热、传质特性[23a,29]。作者目前正在研究这些生产进一步放大的可能性。【公司简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20210922/20210922141810_9943.pdf
应用实例
2021.11.29

【河北工业大学案例】荷兰Chemtrix微反应器中邻氯甲苯催化氧化制备邻氯苯甲醛
【实验背景】邻氯苯甲醛(OCBD)是合成药物、染料和农药的重要中间体。它的的合成方法包括:邻氯苯氯的水解、邻氯甲苯(OCT)的电化学氧化及过氧化氢或氧分子对OCT的直接氧化等。最近有报道称,在CdS-ZnS光催化剂上,以K2S2O8为氧化剂,当存在过量聚甲基氢硅氧烷或分子氧时,OCT也可以发生催化氧化,从而得生成OCBD。然而迄今为止,工业上主要是通过OCT的侧链氯化反应生成邻氯苯氯,再经过水解步骤来制备OCBD。这样的工艺通常过程复杂且造成环境污染。相比之下,分子氧氧化剂具有廉价易得、环境友好及原子经济性高等优点,所以被认为是一种更优的选择。近几十年来,已有很多研究尝试在多种催化剂上对取代甲苯进行直接有氧氧化来制备苯甲醛和苯甲酸。其中,MC催化剂(由Mid-Century公司发现的Co、Mn和Br化合物的组合)已成功被应用于工业间歇反应器中苯甲酸和苯甲醛的生产。但是间歇反应器中该氧化反应存在爆炸的风险。同时,由于氯苯甲醛容易发生进一步氧化而变成酸,所以很难实现氯苯甲醛的高选择性。相比之下,连续流微通道反应器彻底克服了间歇反应器的这些安全及工艺问题,可以最大限度地降低安全风险。同时它允许使用高温和高压条件,从而可以提高氧化效率。近年来,微通道反应器技术在化学领域取得了长足的发展。与传统间歇反应器相比,连续流微通道反应器具有“比表面积大、传热/传质效率高、可操作性强、安全性高、副产物少”的优点,而且可对反应条件进行精细控制。迄今为止,微通道反应器已成功应用于各种有机反应,如:醇醛缩合、迈克尔加成、有机金属试剂的亲核加成、Wittig反应、Suzuki-Miyaura偶联、Diels-Alder加成、羰基化合物的Wolff-Kishner还原及各种底物的有氧氧化反应。其中,Stabl等人采用微反应器对醇和醛进行有氧氧化反应,取得了良好的效果。此外,Gemets等人也报道了许多连续流微反应中有氧氧化的成功案例。基于上述研究现状,河北工业大学张月成老师课题组在Chemtrix B.V.公司Protrix型微通道反应器中,以掺有少量水的乙酸为反应介质,采用MC催化体系对OCT进行选择性有氧氧化。考察了催化剂体系组成、氧/OCT摩尔比、反应温度和压力、停留时间等条件的影响。最终得到了OCT有氧氧化制备OCBD的最佳工艺条件。文章发表在Org. Process Res. Dev. 2020, 24, 2034?2042 (DOI: 10.1021/acs.oprd.0c00135)【实验部分】2.1 试剂和仪器分析级邻氯甲苯、四水乙酸钴(Co(OAc)2?4H2O)、四水乙酸锰(Mn(OAc)2?4 H2O)、溴化钾(KBr)、冰醋酸和工业级纯氧为采购所得,且为直接使用。蒸馏水为实验室自制。在Chemtrix B.V.公司Protrix型碳化硅微通道反应器中进行氧化反应。该反应器连接Bronkhorst公司的气体质量流量计和控制器。采用安捷伦1260 Infinity高效液相色谱仪(装有shimp -pack VP-ODS Ce色谱柱)对反应混合物进行分析。2.2 分子氧氧化OCT将一定量的Co(OAc)2·4H2O、Mn(OAc)2·4H2O和KBr溶解于冰醋酸与水的特定比例混合容剂。再加入一定量的OCT,搅拌至透明均匀。在特定的温度和压力下,将配合好的反应溶液由10 mL进料泵送入微反应器。然后与气体质量流量控制器控制进入的氧进行混合。氧气量可由理想气体定律(式1)求得:式中,P为反应压力(Pa),T为室温(K),V为气体质量流量控制器记录的气体体积(m3), R为通用气体常数(8.314 J/mol?K)。反应结束后,采用高效液相色谱技术对工艺流程进行研究,采用外标法进行定量分析。OCT的转化率及对OCBD和邻氯苯甲酸(OCBA)的选择率(mol %)计算公式如下:其中nOCT,initial和nOCT,converted分别为OCT的初始和转换量(moles)。而nOCBD和nOCBA分别是OCBD和OCBA的生成量(moles)。【 结果与讨论】3.1 KBr用量的影响反应条件:0.158 mol OCT;0.15 mol % Co(OAc)2?4H2O;0.15 mol % Mn(OAc)2?4H2O;47.3 mL乙酸;1.65 wt %水;液体流速= 0.5 mL/min;氧流量= 58 mL/min;停留时间= 9.4 s;反应压力= 0.8 MPa;反应温度= 423 K。总的来说,MC催化剂的催化活性和选择性随Br/(Co+Mn)摩尔比的变化而变化。溴离子作为电子转移引发剂和自由基引发剂,有助于缩短诱导时间,提高氧化速率,是影响反应的关键因素。所以首先研究了溴化钾用量的影响。Co/Mn/Br的摩尔比为0.3/0.3/1。如图1所示,当溴化钾用量从0.05mol%增加到0.35mol%时,OCT的转化率从1.9%增加到了9.5%,OCBA的选择性从8.9%增加到了28.6%。然而,OCBD的选择性从84.2%下降到67.4%。当溴化钾用量介于0.35-0.5 mol%之间时,OCT的转化率有所降低,OCBD和OCBA的选择性变化不大。当采用0.5 mol%溴化钾时,OCT的转化率最高可达11.6%,OCBD和OCBA的选择性分别为65.5%和30.1%,OCBD的收率最高可达7.6%。基于此实验结果,后续实验均使用0.5 mol%的溴化钾。3.2 催化剂用量的影响首先进行了空白实验,包括纯Co(OAc)2·4H2O、纯Mn(OAc)2·4H2O、Co(OAc)2·4H2O加KBr以及Mn(OAc)2·4H2O加KBr的四种催化体系。如表S1-S4所示,这四种空白实验的OCT转换率都非常低。这表明该催化剂体系中的三个组分均不可或缺。反应条件:0.158 mol OCT;47.3 mL乙酸;1.65 wt %水;Co/Mn/Br摩尔比= 0.3/0.3/1;液体流速= 0.5 mL/min;氧通量= 58 mL/min;停留时间= 9.4 s;反应压力= 0.8 MPa;反应温度= 423 K。然后研究了催化剂用量(0.64-0.96 mol %)对OCT氧化反应的影响。Co/Mn/Br的摩尔比为0.3/0.3/1。如图2所示,当催化剂负载量从0.64 mol %增加到0.96 mol %时,OCT的转化率从10.7%增加到了14.4%。同时,OCBD的选择性有所降低,而OCBA选择性有所增加。这说明OCBD发生了进一步氧化,形成了OCBA。为了保证OCBD的选择性,选择负载量为0.88 mol %的催化剂。OCT的转化率为12.9%,OCBD和OCBA的选择性分别为64.3%和31.8%。3.3 氧/底物摩尔比的影响然后研究了氧/底物摩尔比(O/S)的影响。该实验的单位时间内氧通量和反应溶液中底物的浓度保持不变。改变投料比就改变了单位时间内的投料量,从而改变了O/S。如图3所示, OCT的转化率和OCBA的选择性都随O/S的增加而提升,而OCBD的选择性则有所降低。当O/S为1:1时,OCT的转化率仅为3.9%,而OCBD的选择性高达82.7%。当O/S增加到13.8:1时,OCT的转化率和OCBA的选择性分别达到15.8%和39.9%,而OCBD的选择性下降到57.0%。为了实现“单位时间内获得更多OCBD”的目标,他们确定了最佳的O/S为5.5:1。此时,OCT的转化率为10.5%,而OCBD和OCBA的选择率分别为72.4%和23.8%。反应条件:0.88 mol %催化剂;Co/Mn/ Br摩尔比= 0.3/0.3/1;47.3 mL乙酸;1.65 wt %水;氧通量= 58 mL/min;停留时间= 9.4 s;反应压力=0.8 MPa;反应温度= 423 K。3.4 反应温度的影响由于OCT有氧氧化是一个自催化放热反应,因此反应温度的影响需要密切关注。图4显示了393-433K范围内温度对该反应的影响。可见,随着反应温度的升高,OCT的转化率和OCBA的选择性有所增加,而OCBD的选择性明显降低。在393 K下,OCT的转化率仅为1.7%,但OCBD的选择性高达88.2%。当反应温度提高到433 K时,OCT的转化率提高为13.3%,而OCBD的选择性降低到了66.2%。经过综合考虑,确定最佳反应温度为423 K。此时,OCBD的收率可达到7.6%。反应条件:0.158 mol OCT;0.88 mol %催化剂;Co/Mn/Br摩尔比= 0.3/0.3/1;47.3 mL乙酸;1.65 wt %水;液体流速= 1ml /min;氧通量=58 mL/min;停留时间= 9.4 s;反应压力= 0.8 MPa。3.5 氧气压力的影响OCT的氧化是气-液相反应,因此氧压力的提高能促进氧在液相中的溶解。而且加压可以改变微通道反应器中的液体流动状态,从而影响反应的收率和选择性。所以他们又考察了氧压力在0.3~1.0 MPa范围内对目标反应的影响。如图5所示,OCT的转化率几乎保持在10%左右。在0.3-0.6 MPa的低氧压力范围内,OCBD和OCBA选择性的变化趋势有所不同。当氧压为0.6 MPa时,OCBD的选择性最高,OCBA的选择性最低,且二者均随着氧压的进一步升高而保持基本不变。反应条件:0.158 mol oct;0.88 mol %催化剂;Co/Mn/Br摩尔比=0.3/0.3/1;47.3毫升ofacetic酸;1.65 wt %水;氧(或空气)通量=58 mL/min;氧/OCT摩尔比=5.5;停留时间=9.4 s;反应1温度= 423 K。在0.3 MPa的压力下,用空气代替氧气作为氧化剂进行反应。如图5所示,在相同的反应条件下,其OCT的转化率仅为0.2%,远远小于10%。这主要是由于空气中的氧气浓度较低,从而使得液相中的氧气浓度也偏低。最终确定最佳的氧气压力为0.5 MPa。此时,OCBD的选择性达到71.8%,对应的收率为7.4%。3.6 停留时间的影响微通道反应器中的连续反应,反应物的停留时间也是关键影响因素。因此,最后考察了停留时间对反应的影响。如图6所示,OCT的转化率随着停留时间的延长而增加,OCBA的选择性也随之增加。而OCBD的选择性随停留时间的增加而降低。当停留时间大于9.4 s时,OCBD的选择性小于70%。为了尽可能多地获得OCBD,作者将停留时间确定为9.4 s。此时,OCT的转化率达到10.3%,对OCBD和OCBA的选择性分别为71.8%和25.2%。反应条件:0.158 mol oct;0.88 mol %催化剂;Co/Mn/Br摩尔比:0.3/0.3/1;47.3 mL乙酸;1.65 wt %水;氧/OCT摩尔比= 5.5;反应压力= 0.5 MPa;反应温度= 423 K。3.7 最优条件基于上述实验研究结果,作者确定了微通道反应器中OCT有氧氧化合成OCBD的最佳反应条件为:乙酸/H2O混合溶剂(aq,1.65 wt%);0.88 mol%催化剂;Co/Mn/Br摩尔比为0.3:0.3:1;氧/OCT摩尔比为5.5:1;停留时间为9.4秒;反应温度为423 K;氧气压力为0.5 MPa。在此条件下,OCT的转化率为10.3%,OCBD的选择性为71.8%,相应的OCBD产率为7.4%。【正交实验】为验证单因素优化结果的可靠性,他们进一步组织了正交试验。Co/Mn/Br摩尔比为0.3:0.3:1,反应压力为0.5 MPa。表1所示为四因素三水平(34)正交实验表。图7显示了转化率和选择性与四个因素及水平的关系。很明显,这四个因素都随着水平值的增加而发生单调的增加或减少。其中,因素B和C随水平变化的幅度比较大。而且值得注意的是,因子C的转化率和选择性随着反应温度的升高而显著增加或降低。这导致表2中的R值最高。因此,可以认为对转化率和选择性影响最大的因素是反应温度。表2为正交实验的综合平衡分析结果。可见,对于因子A,最高的OCT转化率出现在A3水平,最高OCBD选择率出现在A1水平。那么综合考虑,为了获得尽可能多的OCBD,最终选择为A2水平。同理,B2、C2和D2也分别是因素B、C和D的首选水平。基于以上分析,该研究工作最终确定了微反应器中该反应的最佳工艺为A2B2C2D2,即: Co/Mn/Br摩尔比为0.3:0.3:1;催化剂总量为0.88mol%;反应温度为423K;氧/OCT摩尔比为5.5;反应压力为0.5MPa;停留时间为9.4s。可见,正交实验结果与单因素优化试验结果完全一致。【可行性实验】为了确定该工艺的实际可操作性,收集了最佳反应条件下的反应液(约1000mL),并尝试将其中的OCBD分离出来。首先,将收集的反应液在50℃-0.1Mpa下蒸馏,以除去乙酸和水;然后升温至80℃以除去OCT;再将残余物料在90℃-0.1MPa下蒸馏,即得到了OCBD粗产物。最后,将该粗产物转移到小烧瓶中再次蒸馏,即得到了纯度为98.5%的OCBD。收率为70%。表3将该工作的结果与相关文献进行了比较。虽然该论文报道的OCBD收率微微低于文献报道的间歇反应器中固体催化剂(Co,Mn)2O4及KBr催化的OCBD收率,但是连续流微反应器成功避免了间歇反应器中的安全隐患。因此,从OCT转化率和OCBD选择性及过程安全性的角度综合来看,可以认为该论文研究结果优于其他催化剂体系。最后,将前面得到的最优条件应用于多种其它底物的有氧氧化反应,以制备相应的醛。结果如表4所示。与OCT相比,甲苯和4-叔丁基甲苯的氧化转化率较高,但醛的选择性较低。这主要是由于甲苯和4-叔丁基甲苯的电负性比OCT强(表4,第1-2条)。当以OCT、对氯甲苯和间氯甲苯为底物时,反应结果也十分类似。醛类产物的选择性在70%-72%范围内(表4,第3-5条)。2-甲基吡啶氧化的转化率和醛选择性均低于OCT。这表明该催化体系和最佳条件与甲基杂环芳烃不太相容(表4,第6条)。【结论】总之,该工作以乙酸掺少量水为溶剂,以Co(OAc)2/Mn(OAc)2/KBr为催化剂体系,在微通道反应器中进行OCT选择性有氧氧化来制备OCBD;考察了催化剂负载量、催化剂组成、氧/OCT摩尔比、反应温度、反应压力和停留时间等因素对反应的影响,确定了最佳反应条件。而且结果显示,维持OCT的低转化率对高选择性靶向制备OCBD至关重要。在最佳反应条件下,OCT转化率为10.3%,OCBD的选择性达到71.8%。原文:Catalytic Oxidation of o-Chlorotoluene with Oxygen to o-Chlorobenzaldehyde in a Microchannel Reactor. Organic Process Research & Development[J]. 2020, 24(10): 2034-2042【公司简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20211020/20211020134733_0539.pdf
应用实例
2021.11.29

【复旦案例】维生素B1关键中间体2-甲基-4-氨基-5-氨甲基嘧啶的全连续流合成
【背景介绍】甲基-4-氨基-5-氨甲基嘧啶(2)是生产维生素B1(1)(图1)的关键中间体。1维生素B1对维持人和动物的神经传导、心脏和胃肠道的正常功能是必不可少的。2,3根据《2020年全球维生素B1(盐酸硫胺)市场》研究报告显示,2020年全球维生素B1市场收入为6.488亿美元预计到2026年将达到8.547亿美元。3到目前为止,已经有大量的工作投入到维生素B1的合成中,分别采用了不同的合成策略。然而,通过关键中间体2-甲基-4-氨基-5-氨甲基嘧啶(2)作为母核仍然是工业应用中的一种可靠的方法。2中间体2的合成已在大量文献中进行了描述,在此作者针对包括之前在间歇操作中的研究成果在内的四个策略进行了讨论,4-19总体比较结果见表1。图1. 维生素B1分子结构表1. 关键中间体2的合成策略总结上述总结的釜式方法均在一定程度上存在反应时间长、副产物生成、后续纯化步骤繁琐等缺点。这些问题大多是使用传统的间歇式工艺技术所固有的。众所周知,连续流合成技术能够克服间歇式工艺固有的一些缺陷,在更好的传质和传热、良好的极端反应条件和危险控制、较低的工厂占地面积和生产灵活性等方面显示出独特的优势。20-22连续流技术通过避免中间提纯和加工工序,可以实现反应的质量控制,实现快速生产和环保安全。在改进质量控制、降低成本、提高工艺安全性和显著缩短时间等方面的潜力推动下,创新性地采用连续流技术对工业的未来至关重要。23-26路线1. 2-甲基-4-氨基-5-氨甲基嘧啶(2)的合成路线复旦大学手性分子工程中心的陈芬儿院士课题组采用了氰乙酰胺方法(路线1)在连续流系统中高效且经济地合成了4-氨基-5-氨基甲基-2-甲基嘧啶(2),展示了一种快速全连续流动化学生产原料药的高效工艺方法。文章发表在Org. Process Res. Dev. 2021, 25, 10, 2331–2337(https://doi.org/10.1021/acs.oprd.1c00253)。【实验方法】步骤1:合成2-(二甲氨基亚甲基)丙二腈(4)。首先将氰乙酰胺(5)、吡啶溶于DMF中。然后在10ml聚四氟乙烯盘管反应器(内径0.8mm)中用POCl3处理混合物(图2)。聚四氟乙烯的两部分盘管分别保持在0℃ 和25℃。该实验考察了温度、压力和滞留时间对反应结果的影响。样品经淬灭和DCM萃取后,进行GC/MS分析,在最佳条件下采集约10mL样品,中和萃取后,真空除去DCM和过量的DMF,最终得到了黄色油状的2-(二甲氨基亚甲基)丙二腈(4)(产率94%,GC/MS纯度97%,含有2.8%的DMF)1H NMR(400 MHz,DMSO-d6)δ 7.65(s,1H)、3.19(s,3H)、3.16(s,3H);13C核磁共振(101兆赫,DMSO)δ 159.0, 118.2, 116.5, 46.8, 37.9; m/z(GC-MS):148、113、86、51;GC/MS保留时间:4,8.65分钟;DMF,2.55分钟。图2. 2-(二甲氨基亚甲基)丙二腈(4)的连续流合成DMF溶液与POCl3溶液接触后发生Wilsmerele反应,产生强烈的热量。放热反应对间歇式反应器的放大带来了很大的挑战,然而,在将这种合成转化为连续流动过程时,没有观察到严重的温度升高或压力升高现象,安全性较好。同样,一些研究小组也报道了由于微通道反应器中的有效传热而改善的流动反应情况。21在该反应工艺设计中,本文采用了两个微通道盘管反应器分别保持在0℃ 和25℃的温度下进行。在作者的初步研究中,在12.6 ml SiC Protrix反应器(Chemtrix.B.V.)中,用POCl3和吡啶在25℃下处理预溶于DMF中的氰乙酰胺(5)。在30分钟的滞留时间后,原料的转化率仅为92%,收率也较低(78%)(见附录表S1和S2)。当延长一倍停留时间或改变反应温度时,都不能提高反应的转化率。另外,通过GC/MS分析,化合物5由于与过量DMF和POCl3反应过度而转化为环状氯化杂质(化合物7见路线2)。而由于5在DMF中的溶解度较差,本文在40℃水浴条件下将吡啶和化合物5充分溶解在DMF中。考虑到其为放热反应,因此将混合物泵送至与POCl3在2 ml PTFE盘管反应器中接触反应时,温度为0℃。然后再连接一个8ml的盘管反应器,在25℃的水下继续进行反应。实验发现在18分钟的滞留时间内完成了原料的完全转化。接着本文进行了全面的研究,以探讨优化连续流反应条件(表2)。表S1: 在Protrix碳化硅微通道反应器中实现2. 2-(二甲氨基亚甲基)丙二腈的连续流合成图S2. 组分4 的Protrix碳化硅微通道反应器(荷兰Chemtrix BV)连续合成配表2. 不同条件下流动合成2-(二甲氨基亚甲基)丙二腈(4)的结果表2. 不同条件下流动合成2-(二甲氨基亚甲基)丙二腈(4)的结果经过初步实验发现,在25℃无压力的条件下,使用Protrix微通道连续流反应器可在18分钟的停留时间内实现原料5的完全转换(表2,entry 3)。将盘管2的温度升高到30和40℃ 得到了相同的结果,但收率降低,产生了更多杂质(表2,entry 1和2)。另一方面,将盘管2的温度降低到15℃时,化合物5没有充分反应(表2,entry 5和6)。同时,当反应背压提高时,原料的转化率略有增加。在将停留时间缩短到12min后,原料几乎完全转化,收率较高(表2,entry 7和8)。当采用0和25℃ 同时带有3 bar背压的条件下,实验得到最佳结果,得到的2-(二甲氨基亚甲基)丙二腈(2)产品的分离产率为94.7%,纯度99.5%。与Zhao等人在-10℃条件下的12h间歇过程(产量为74%)19相比,这种连续流程更有效更安全,也更易于工业放大(表3)。在Zhao等人的研究过程中19发现有两个杂质化合物6和7,如路线2所示。他们认为,杂质6容易在溶液的pH值大于3时生成,并且会在最终分离过程中被除去,而杂质7容易在高温条件下生成,并且在加入一定量的吡啶后杂质7含量降至最低。然而,在本文的连续流合成研究中,这两种杂质均没有被观测到,该结论得到了HPLC和NMR的验证。在微通道连续流反应器中通过对温度和混合淬灭等过程的良好稳定的控制,实现了高效的传质传热效果。在此基础上,PTFE管路可以直接进行线性放大从而实现千克级的生产27,同时连续流生产过程更加的简易安全。表3. 间歇釜式和连续流合成2-(二甲氨基亚甲基)丙二腈(4)的工艺比较步骤2:合成2-甲基-4-氨基-5-氰基嘧啶(3)。接下来继续进行第二步反应,2-(二甲氨基亚甲基)丙二腈(4)在甲醇中与乙脒-甲醇溶液进行混合,反应得到化合物3沉淀,该过程在连续流动中进行了探索优化(图3和表4)。将盐酸乙脒与甲醇钠按1:1.1的摩尔比在甲醇中混合,过滤混合物,收集滤液即得到乙脒的甲醇溶液。在微通道连续流盘管反应器中,沉淀的形成是一个巨大的挑战,必然会出现堵塞现象,这将会导致压力的升高和反应过程的中断。因此,针对该反应,本文采用了Coflore ACR连续多级搅拌反应器(AM Technology )来避免任何可能由沉淀形成引起的问题。具体操作如下:在Coflore ACR连续多级搅拌反应器(90 ml反应体积)中用乙脒甲醇溶液处理甲醇中的2-(二甲氨基亚甲基)丙二腈(4)。实验考察了温度、浓度、停留时间和震荡频率对反应结果的影响。将反应料浆过滤并溶解于甲醇中,然后通过GC/MS进行分析。为了进行光谱表征,在最佳条件下收集约50 mL样品,过滤并真空干燥后得到白色固体形式的2-甲基-4-氨基-5-氰基嘧啶(3)(产率90%,GC/MS纯度99%)。1H-NMR(DMSO,400MHz):8.50(s,1H),7.29(br s,2H),2.57(s,1H);m/z(GC-MS),134(m+),94,66;GC/MS保留时间:3,6.73分钟。图3.连续流合成2-甲基-4-氨基-5-氰基嘧啶(3)英国AM Technology 连续多级搅拌反应器ACR图片图S3 Coflore ACR反应器中2-甲基-4-氨基-5-氰基嘧啶的连续合成造成的堵塞(大量沉淀物)表4. 不同条件下流动合成化合物3的结果体" >甲基-4-氨基-5-氰基嘧啶的连续合成造成的堵塞(大量沉淀物)如表4所示,使用2-(二甲氨基亚甲基)丙二腈(4)和乙脒进行反应,原料的转化率随着温度和频率的增加而增加。然而,由于产物2-甲基-4-氨基-5-氰基嘧啶(3)在甲醇中的溶解度很低,浆料中沉淀的形成在很大程度上取决于原料2的浓度。实验发现,在ACR中,当滞留时间较长时,1.75 M的化合物2与乙脒(1.1当量)反应会造成反应器的堵塞(附录图S3)(表4,1-3)。虽然减少停留时间有助于缓解堵塞,但结果同样不令人满意(表4,4&5)。然而,在改进的混合条件下(5Hz提高到6Hz),原料4浓度的略微降低会引起化合物3转化率的增加(表4,6-11)。而其浓度进一步降低会导致反应时间的延长,从而实现化合物2完全转化(表4,12-13)。因此,最终实验确定了最佳工艺条件为55℃,频率6 Hz,25-30min滞留时间,化合物4的浓度为1.25M,反应样品通过GC/MS检测发现几乎完全转化为3,分离收率为90%。同样,该连续流合成工艺结果3优于之前间歇釜式报道的结果19(表5)。这再次证明了连续流合成工艺在传质传热和控制反应条件方面具有显著的优势,克服了釜式反应反应时间过长的缺点。表5. 间歇釜式和连续流合成3的结果比较步骤3:2-甲基-4-氨基-5-氨甲基嘧啶(2)的合成。2-甲基-4-氨基-5-氰基嘧啶(3)的后续氢化反应过程通过固定床反应器来(深圳一正科技有限公司,20 mm/100 mm,填充12.4 g改性雷尼镍,2.2 ml反应体积)完成(图4)。2-甲基-4-氨基-5-氰基嘧啶(3)在甲醇中通过高压泵送入填充改性雷尼镍催化剂的固定床反应器中,并在100℃下与氢气进行反应,体系背压为16bar。实验考察了滞留时间和反应物浓度对反应结果的影响。从气液分离器出口采集样品,用高效液相色谱法进行分析。为了进行光谱表征,在最佳条件下收集了约20ml样品,甲醇溶剂在真空中被去除,产物为类白色固体(产率99%,HPLC纯度>99%)。1H NMR(二甲基亚砜,400Hz):δ=2.28(s,5H)、3.53(s,2H)、6.67(s,2H)、7.88(s,1H);质谱:139[M+1]+;HPLC保留时间:4, 1.22分钟。图4. 连续流合成2-甲基-4-氨基-5-氨甲基嘧啶(2)span>微反应固定床反应器(深圳市一正科技有限公司)图片表6. 不同条件下流动合成化合物2的结果化合物3和碱催化剂由泵输送进固定床反应器中,反应温度保持在100℃。实验发现(表6),在1-2分钟的滞留时间内,原料3完全转化(表6,1-2)。当增加液体流速时,反应时间缩短,同时转化率降低(表6,3-6),而气体流速的增加和浓度的降低对转化率没有明显的影响。此外,氨水和三乙胺在加氢反应中都可以作为碱性催化剂使用。该反应的最佳条件为100℃,停留时间为1.5-2min,此条件下产物2的收率定量,纯度>99%。间歇式和流动化学合成2-甲基-4-氨基-5-氨甲基嘧啶(2)的工艺比较如表7所示。连续流工艺下的空间产率(2.23moL/L/h,即307 g/L/h)是间歇式的15倍(0.15 moL/L/h,即 20.7 g/L/h)。值得注意的是,考虑到生产规模,本文中的生产效率仅为0.15 g/h,比釜式生产效率(0.41g/h)低2.7倍。然而,由于连续流工艺在重复试验中体现出的稳定的生产效率,生产规模可以通过进行多模块并行的方式进行放大生产。表7. 间歇釜式和连续流合成2的结果比较全连续合成工艺流程。当确定了各个步骤的最佳条件后,在此基础上,作者继续将它们组合成一个完全连续的过程(图5)。应注意的是,本文中的完全连续过程指的是无额外中断的连续生产过程,尽管使用了缓冲罐用于中间体的储存、溶剂转化和重新溶解。所有工艺均以连续或至少多步连续的方式进行。这参考了Cole等人在连续流动条件下以千克规模合成单乳酸酯的工作,27以及作者先前报道的一种合成3-氯-4-氧戊烯乙酸酯的完全连续过程。28图5.全连续合成2-甲基-4-氨基-5-氨甲基嘧啶(2)工艺流程图化合物2的连续合成在三个流动单元中进行,包括六个连接的流流设备。流动的第一单元包括氰乙酰胺(5)和吡啶的DMF溶液,在0℃ 和25℃ 和3bar的背压下与三氯氧磷进行反应,时间为18分钟。随后反应溶液采用用过量的20 %的氢氧化钠溶液进行淬灭。然后将流出物收集在缓冲容器中,并在室温下通过DCM在CINC环隙分离萃取器中进行高效萃取(图S7)。其中,有机相直接在泵入连续旋蒸设备,在45℃条件下去除二氯甲烷,并加入甲醇,得到化合物4的甲醇溶液。将该溶液泵入流动第二单元,与即时制备的乙脒甲醇溶液进行反应。流动第二主要包括ACR连续多级搅拌反应器,温度保持在55°C,反应时间约30分钟。反应产物随后进入连续过滤器。滤液去除后,用甲醇溶解滤饼。流动第三单元包括一个固定床反应器,填充了改性雷诺镍,反应温度保持在100℃,反应压力为16 bar,反应时间为2分钟。最终实验以84%的产率得到化合物4,总停留时间为74分钟,生产效率为0.92g/h。综上,维生素B1母核2-甲基-4-氨基-5-氨甲基嘧啶(2)的全连续流合成工艺改善间歇釜式生产中的反应条件,提高了安全性,节约成本。采用德国CINC环隙离心萃取器进行连续分离【结论】1、选用合适的流动设备进行单步连续流合成实验,探讨不同反应因素的影响从而确定最佳的合成条件。2、在流程中可加入连续淬灭和萃取过程,并在进行下一步反应前浓缩所需化合物。该工艺不需要进行额外的中间纯化和分离的过程。3、本研究以氰乙酰胺为原料,采用全连续流动化学合成工艺在6个连续流动设备中完成了3次化学转化过程,成功地合成了维生素B1的关键中间体2-甲基-4-基-5-氨甲基嘧啶(2)。产品收率为84%,总反应时间为74分钟。【原文】Org. Process Res. Dev. 2021, 25, 10, 2331–2337(https://doi.org/10.1021/acs.oprd.1c00253)【一正科技简介】深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司连续多级搅拌反应器、英国Autichem催化加氢系统、Creaflow连续流光化学反应器、瑞典Spinchem公司等在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。公司网址:www.e-zheng.com联系电话:0755-83549661产品热线:400-0755-403应用原文及附件请下载下方PDF文件:/include/upload/kind/file/20211112/20211112145155_9487.pdf/include/upload/kind/file/20211112/20211112145257_2964.pdf
新品
2021.11.26

【复旦大学案例】维生素B1重要中间体3-氯代-4-氧代乙酸戊酯的全连续流合成
【背景介绍】3-氯代-4-氧代乙酸戊酯(2)是工业生产中合成维生素B1(1)的一个关键中间体。1维生素B1(图1)作为一种重要的基础营养成分,在碳水化合物和支链氨基酸的代谢中起着重要作用,特别是对人类和动物的生长和健康。它能够保护神经系统,促进肠胃蠕动,帮助碳水化合物笑话,改善精神状况,维持神经系统、肌肉、心脏的正常活动,同时有助于带状疱疹的治疗。2根据《2020年全球维生素B1(盐酸硫胺)市场》研究报告显示,2020年全球维生素B1市场收入为6.488亿美元预计到2026年将达到8.547亿美元。3图1. 维生素B1化学结构近二十年来,流动化学以及连续工艺受到了学术和工业界的广泛关注和认可,一些在传统釜式方法无法或者极难实现的化学反应和技术流程可以通过连续流的方式得到改进。连续流技术在传质和传热方面显示出独特的优势,能够很好地控制一些极端反应条件,并具有占地面积和生产灵活性等优点。在一些简化系统和高自动化平台均可进行活性药物成分的合成研究,连续流工艺使很多工艺过程变得更加安全和有利于生产的扩大化。4-6在提高质量控制、降低成本、提高工艺安全性和显著缩短批量操作时间的潜力推动下,创新性地采用连续流技术对未来的工业界至关重要。7-10尽管维生素B1的合成目前已经被认为是一种较成熟的生产方法,但仍需要通过连续流动合成技术进行改进,以实现更经济、更安全和更绿色的愿景。文献中报道了关于维生素B1重要中间体3-氯代-4-氧代乙酸戊酯(2)的间歇釜式合成路线(参见路线1)。美国专利29326531111在1960年报道了一种方法,将乙酰丁内酯与无水乙酸钠和冰醋酸混合,用氯气进行氯化。然后在真空蒸馏的条件下,向含有乙酸的滤液中加入浓盐酸和醋酸酐,从而制得化合物2。1981年Plazzi,121978年Archer和Perianayagam13以乙酰丁内酯(4)和硫酰氯为起始原料,经醋酐酰化得到了总收率低于80%的3-氯代-4-氧代乙酸戊酯(2)。类似地,Hawksley14在2001年报告了乙酰丁内酯(4)用硫酰氯氯化,然后用乙酸和盐酸加热脱羧和乙酰化,然后添加醋酐,最终得到总收率为76%的目标产物。另外,在相同的回流条件下,2008年的Thomas15、2011年的Herbert16和2016年的Nemeri17也报告了使用硫酰氯生成化合物2,然后通过在乙酸中与醋酸酐回流开环和酰化。纯化后的产物收率分别为81%、77%和63%。路线1. 间歇釜式生产3-氯代-4-氧代乙酸戊酯(2)的合成路线上述工艺均为间歇操作,由于传热传质性能差,不可避免地要进行大量的后处理工作。有些反应持续数小时到数天,这是相当费时费力的。复旦大学陈芬儿院士课题组姜博士等人设计了一条全连续流化学合成3-氯代-4-氧代乙酸戊酯的工艺。与传统的间歇釜式过程相比,连续流动工艺中没有中间产物的分离过程,反应温度适中,安全隐患显著降低。同时,连续流动合成与在线萃取分离相结合大大提高了生产率,显著地缩短了时间(从几小时缩短到几分钟)。本文设计的全连续流工艺总的平均停留时间约为32分钟,得到3-氯代-4-氧代乙酸戊酯产品的总产率为90%,纯度为96%,生产效率为1.79 g/h。文章发表在Org. Process Res. Dev. 2021, 25, 9, 2020–2028(https://doi.org/10.1021/acs.oprd.1c00065)作者采用氯气对乙酰丁内酯(4)进行氯化,除醋酸外,第二步引入醋酐进一步脱羧酰化。这是一条全连续的3-氯代-4-氧代乙酸戊酯(2)的生产工艺,提供了一条高效的流动合成路线和连续操作序列,并得到良好的收率和纯度。【实验方法与步骤】路线2清楚地说明了反应的不同阶段和关键设备。路线2. 反应阶段及关键设备 Protrix是一个模块化的碳化硅微通道反应器,通常用于反应条件的筛选和小规模生产实验(参见图S1)。它包含3个SiC模块,可灵活配置以提供所需数量的原料进口、反应体积和温度控制区域。每个模块包含反应通道和集成热交换层,以提供最佳的热交换控制(见图S2)。图S1:Protrix碳化硅微通道反应器(荷兰Chemtrix BV.)连续混合萃取/分离器是由三块PTFE板(分别设计为A、B、C)组成,中间夹有一个2000目不锈钢滤网和一个PTFE膜(孔径为0.45μm)(见图2)。该连续萃取分离设备是由Zefluor膜分离器18,19改进而来,在每个板上蚀刻微型通道,使流体以U字形模式通过微通道(详细示意图见图S4)。 图2. 连续液液膜萃取/分离器 第1步:连续氯化反应。氯化装置如图3所示。Cl2气体经过缓冲瓶和充满浓硫酸的干燥瓶后,通过质量流量计输送入Protrix反应器中,以干燥氯气并防止回流影响。每次反应前都对质量流量计(图6-12)进行校准,以确保使用电子皂膜流量计(GGL-103A,上海红科)获得准确的Cl2气体含量值(详情见附录中的图S5)。液体试剂(化合物4)由柱塞泵输送。采用的Protrix反应器总反应体积为5ml,反应器出口连接气液分离器(深圳市一正科技有限公司提供),分离器上的尾气出口与可调背压调节器相连, 同时在此处引入氮气,从而辅助系统将压力保持在0.5-2 bar的可调范围内。反应过程的温度控制是通过Viar温度控制器来实现的。如图3所示,在Protrix反应器内,进行了一系列的实验。本文设定了两个反应温度(0℃ 和25℃),将化合物11的流速设定在0.5~2 ml/min范围变化,并根据与化合物11的当量比(1:1.0~1:1.5)调节Cl2的流速。图3. α-乙酰基-α-氯代-γ-丁内酯(3)的连续流动合成第2步:连续脱羧/酰化反应。在特定的温度和背压下,保持一定的反应时间,化合物3与醋酸在聚四氟乙烯盘管反应器(内径=0.8mm)中进行混合反应。然后,保持温度和压力条件不变,将反应混合物与醋酐液体泵入到第二个PTFE盘管反应器(内径=0.8 mm)中进行进一步的酰化反应。两个盘管反应器都通过浸在油浴中以控制反应温度。连续萃取分离。在室温条件下,第2步连续流设备的酰化反应产物通过T型混合器引入饱和碳酸氢钠水溶液进行混合和猝灭,然后将混合溶液输送入连续膜萃取和分离器中用二氯甲烷进行萃取。饱和碳酸氢钠溶液和二氯甲烷的流速根据第2步出口的流速进行确定。混合溶液经过减压浓缩后(,45℃), 即可得到目标化合物3-氯代-4-氧代乙酸戊酯(2)。CFD模拟。本文采用了K-Epsilon [2equn] (k-ε model)模型。使用GAMBIT软件(ANSYS)为每种类型的混合器创建三维网格文件。模拟参数直接采用作者在实验过程中的数据。最终模拟计算了T型混合器和十字交叉混合器的入口和出口的质量流量,差值为6.67×10-6和-7.57×10-5 g/s,满足了仿真的要求。图4所示为T型混合器模型(图4a)和十字交叉混合器模型(图4b)的三维图形。 图4.混合器的仿真模型:(a)T型混合器的三维图形建模;(b)十字交叉混合器的三维图形建模【结果与讨论】实验通过对连续流动合成3-氯代-4-氧代乙酸戊酯(2)的体系中,单步连续流反应的的反应当量比、流速、反应温度、滞留时间、压力等因素进行了综合考察。在稳定状态下,每种条件都保持运行了2-5次,以便保证实验数据的可靠性。步骤1:氯化反应实验得到Cl2气体流量计的标定曲线为:实际流速(ml/min)=设定流速(ml/min)*0.4953-0.6619,R2值为1,显示出了良好的线性对应关系(见图S6)。图5显示了不同Cl2和乙酰丁内酯(4)的摩尔比和不同流速下连续流氯化反应的实验转化率结果。图5. 不同条件下连续流氯化反应的转化率结果起始化合物乙酰丁内酯(4)的转化率随当量比(Cl2:(4))的增大而增大。当Cl2与化合物4的摩尔比为1.1~1.5时,转化率达到最大值(100%)。然而,当摩尔比低于1.1时,尤其是在低温下,会出现波动。例如,化合物4的转化率在25℃时,液体流速为1.0ml/min时不断增加,而以相同的流速在0℃时,转化率先降低后升高。此外,在相同温度下,较低流速下反应的波动更为明显。同样的条件运行三次,得到了同样的结果。据推测,这是由于高流速下流动中的有效混合产生了更多的扩散涡流。18,19然而,图5中没有观察到化合物11在相同流速下转化率随温度变化的规律性趋势,也没有观察到化合物11在相同温度下不同流速下的转化率随温度变化的规律性趋势。这是因为液相在气相中的分散受气液流量的影响,特别是气体流量受温度的影响更大。20,21由于流动合成的主要目的是以一种高效、简便的方法获得产物,因此对于该步氯化反应,最佳的反应条件为:室温(25℃),乙酰丁内酯(4)的流速为2ml/min,流动反应的停留时间约为30s。这样,化合物 α-乙酰基-α-氯代-γ-丁内酯(3)的产率为93%,纯度为98%。步骤2:脱羧/酰化反应接下来作者考察了α-乙酰基-α-氯代-γ-丁内酯(3)在连续流动中的脱羧/酰化反应。连续流反应流程如图6所示。两个反应原料(化合物3和醋酸和盐酸溶液的混合物)分别通过两个注射泵通过T型混合器(PTFE,i.d.=2.5 mm)引入5 ml PTFE盘管反应器中(i.d.=0.8 mm)。乙酸和 α-乙酰基-α-氯代-γ-丁内酯(3)的浓度与其在间歇釜式反应中的比例一致(见附录)。反应温度设定为115℃,压力7 bar,停留时间约为15分钟。最终得到 α-乙酰基-α-氯代-γ-丁内酯(3)的转化率为74-78%,反应混合物中化合物5的转化率约为21-26%(筛选实验见表S3)。图6. α-乙酰基-α-氯代-γ-丁内酯(3)的连续流脱羧/酰化反应为了提高化合物2的产率,然后使用T型混合器(PTFE,内径=2.5 mm)将粗产物通过第二个反应器盘管(5 ml,内径=0.8 mm)与醋酐流在115℃下混合反应,压力为7 bar。在反应20分钟后,分离得到3-氯代-4-氧代乙酸戊酯(2),收率为85%,产品纯度93.25%(见表S4)。该结果明显优于作者之前的间歇釜式操作结果,即化合物13的产率为60%,纯度为95%(粗品的产率为63%,纯度为92%)。在间歇过程中,本文将化合物3(1.0equiv)与乙酸(3.2equiv)、水(1.1equiv)、盐酸(35%,0.1equiv)和醋酸酐(2.0equiv)混合,在120℃条件下回流约6小时, 然后进行减压蒸馏得到产品(操作步骤见附录S5)。另外,文献16,17中报道的间歇釜式实验也显示3-氯代-4-氧代乙酸戊酯(2)的反应时间为数天,且收率仅为60-77%。相对于间歇式合成工艺,连续流动合成可以通过提高混合效率、改善传热传质和提高反应选择性,从而显著减少反应时间和成本,从而比间歇操作产生更优的结果,例如流动合成法反应的选择性好,效率高。在此基础上,反应条件进一步进行改进,本文通过提高反应温度、延长反应时间和改变反应当量比,对反应结果进行了研究,从而期望降低杂质的含量,提高合成转化效果。在不同条件下的实验结果如表1和表2所示。表1. 在不同当量和流速下,化合物3与乙酸在第一个流动盘管反应器中连续流动脱羧/酰化的转化结果F1——化合物3流速(ml/min)F2——乙酸混合溶液流速(ml/min) 表2. 在第二个盘管反应器中加入乙酸酐后进一步酰化反应的结果F3——盘管反应器1出口流速 (ml/min)F4——醋酸酐流速(ml/min)流量F1-F4是由反应时间和反应物之间的当量比计算决定的。值得注意的是,化合物3在125℃时第一个盘管反应器中就已实现约90%的转化率(表1),此时的反应时间为25分钟。根据表2可知,最终化合物3-氯代-4-氧代乙酸戊酯(2)以90%产率和>95%的平均纯度由第二个连续流盘管反应器直接得到。此结果表明,与图6所示的反应流程相比,随着反应温度的升高和反应时间的延长,大部分化合物5已转化为化合物2。因此,本文最终确定的连续流有机合成3-氯代-4-氧代乙酸戊酯(2)(见图7)的最佳条件如表3所示。图7. 连续流动化学合成3-氯代-4-氧代乙酸戊酯(3)的工艺表3. 全连续流动合成3-氯代-4-氧代乙酸戊酯(2)工艺最佳条件*压力在7~10bar范围内时,结果变化不明显 在以上连续流改进工艺的基础上,本文考虑到在第一步脱羧/酰化过程中获得的较高转化率,决定简化合成路线,去除醋酸酐的添加步骤,如路线3所示。路线3. 全连续流动合成3-氯代-4-氧代乙酸戊酯(2)的改进合成路线另外,为了进一步改进连续脱羧/酰化过程,除了考虑反应当量、反应温度和反应时间等因素外,作者还考虑了混合条件的优化。十字交叉混合器优于T型混合器的特性已经由其他微流研究人员通过实验和模拟结果进行了讨论。18-20因此,本文中十字交叉混合器(PTFE,内径=2.5 mm)被用于增强两相的液-液混合过程(图8)。图8.使用十字交叉混合器改进3-氯代-4-氧代乙酸戊酯(3)的连续脱羧/酰化过程在图8的改进工艺中,α-乙酰基-α-氯代-γ-丁内酯(3)液体作为单独的进料泵入十字交叉混合器,而乙酸和盐酸的混合水溶液则通过T形接头被分成两股支流。因此,一共有三股流体同时被泵入交叉微混合器中进行混合。脱羧/酰化反应选用新的反应器盘管(10ml,内径=0.8mm)进行,反应温度通过油浴控制在125℃,平均滞留时间为30分钟左右,系统背压7-10 bar。在最佳条件下,本文得到3-氯代-4-氧代乙酸戊酯(2)的收率为89-91%,纯度为96.7% (试验结果见表4)。表4. 用4 equiv的乙酸在十字交叉混合器中进行连续流脱羧/酰化反应生成化合物2的条件试验F1——化合物3流速(μl/min)F2——乙酸混合溶液流速(μl/min)图9. 使用T型混合器和十字交叉混合器在相同流速下进行重复反应的结果对比 (F1=22.0 μl/min, F2=49.4 μl/min)如图9所示,使用十字交叉混合器可显著提高反应效率。虽然已有多篇文献报道了对称结构18-21具有较高的混合效率,但在不同的反应体系中,混合条件随流速的不同而发生改变。因此,由于交叉混合器而导致的产品转化率升高的这种现象是值得研究的。在该项工作中,体系的总流速较低(雷诺数为3.6),这是以前没有报道过的。此外,微混合器内部的密度和速度分布很难测量。因此,作者采用了计算流体力学(CFD)来模拟T型混合器和十字交叉混合器中的混合状态,以便更好地了解微观混合过程(图10)。图10. (a)T型混合器中两相混合的速率分布; (b) T型混合器中两相混合的密度分布; (c)十字型混合器中两相混合的速率分布; (d)十字型混合器中两相混合的密度分布如图10a和10c所示的结果表明了十字交叉混合器比T型混合器能够更好更均匀的速度分布。同时类似结果这也体现在了图10b和10d中,图10b和10d显示出了化合物3液体和醋酸水溶液在两个微混合系统中的密度分布。在交叉混合器中反应物在初混合的位置就可以获得稳定的密度分布,而在T型混合器中,在混合区域中更宽,而且体现出了一种较不稳定的状态。这一结果与Luo课题租22,23的研究结果是一致的:在低雷诺数()的情况下,对称结构对两相流的混合更为有效。这是因为在十字交叉混合器中,化合物12的液体流可同时与管流中来自两侧的乙酸溶液进行混合。然而,在T型混合器中,流体由于压力作用在管道一侧附近流动,只在另一侧的有限区域与酸溶液接触(如图10a和10b所示)。此外,管壁附近的低流速也影响了分子间的扩散效率,不利于充分混合。这可以用雷诺数方程Re=dρν/μ来解释,其中,d是盘管直径(m),ρ 是流体的密度(kg/m3),ν 是流速(m/s),μ 是粘度系数(Pa·s)。由于两种混合器的直径和粘度相同,管内对流的混合主要受速度和密度分布的影响。这在一定程度上验证了在低雷诺数的情况下,几何结构对混合性能的影响。全连续流动合成工艺图11显示了3-氯代-4-氧代乙酸戊酯(2)的一条完全连续流动合成的工艺流程,具有比间歇釜式更显著的优势。同时,作者将连续流动合成与连续淬灭、在线连续萃分离取两个步骤结合起来进行操作,具体全连续工艺操作条件见表5。图11. 3-氯代-4-氧代乙酸戊酯(2)的全连续流动合成工艺流程图表5. 全连续流动合成3-氯代-4-氧代乙酸戊酯(2)的操作条件在连续膜萃取分离器的出口可以得到目标化合物2的有机溶液。经过减压蒸馏后,即得到3-氯-4-氧代戊基乙酸酯(2)产品,总收率为90%,纯度96%。该全连续工艺的生产效率约为1.79 g/h,主要由最后一步脱羧/酰化反应的产率控制。不过,生产中可以通过运行并联连续流动设备的方法来进行放大生产。图11的工艺可以并已成功运行12小时,分离出约19.1g产品。因为产品的纯度与间歇式反应纯化后的产品相近(表6),因此连续流的粗产品可以直接用于下一步维生素B1的生产。如表6所示,本研究所进行的3-氯代-4-氧代乙酸戊酯(2)的连续流合成工艺与间歇式操作相比,有效地缩短了反应时间,提高了生产效率,可以为今后的工业生产带来显著的经济效益和市场价值。表6. 间歇釜式与全连续流工艺结果对比【结论】l 通过对单步连续合成反应的实验条件筛选,本文首次成功地开发了3-氯代-4-氧代乙酸戊酯(2)的全连续流化学合成工艺。l 与传统的间歇釜式过程相比,本研究在连续流动化学操作方面取得了一些进展:连续流动工艺中没有中间产物的分离过程,反应温度适中,安全隐患显著降低;l 通过CFD模拟研究,本文采用十字交叉混合器改善了系统的微混合状态,同时不需要乙酸酐进一步酰化;连续流动合成与在线萃取分离相结合大大提高了生产率,显著地缩短了时间(从几小时缩短到几分钟)。l 最后,全连续流工艺总的平均停留时间约为32分钟,得到了3-氯代-4-氧代乙酸戊酯(2)产品,总产率为90%,纯度为96%,总的生产效率为1.79 g/h。此全连续合成工艺可以并已经平稳运行12小时,生成了约19.1 g目标化合物。l 生产的放大可以很容易地通过一定数量的流动反应设备并联实现。预计在将来,全连续流动工艺可普遍应用于工业生产维生素B1或其他药物活性中间体。【原文】Org. Process Res. Dev. 2021, 25, 9, 2020–2028Organic Process Research & Development Article ASAPDOI: 10.1021/acs.oprd.1c00065 荷兰Chemtrix 公司简介:Chemtrix BV. 公司一直致力于研究实验室化工厂——用于制药和化学工业的研究和生产的流动化学合成系统,拥有从应用于研发阶段的低通量微通道反应器(Labtrix Start,Labtrix S1)到生产阶段的高通量微通道反应装置(3D-Print,KiloFlow,Protrix,Plantrix),让使用者可以实现从研发到生产的直接跳跃。Chemtrix BV.公司是世界领先的连续流化学合成技术和连续流动化学合成仪器的供应商,在流动化学领域拥有多项专利技术,公司拥有世界一流的应用研究团队,总部设在荷兰,应用研发部门Chemtrix R&D实验室设在英国郝尔大学。由于公司强大的技术支持,Chemtrix除了提供流动化学反应系统产品以外,还提供以下服务:1、合同研究、研发外包2、化学可行性研究3、工艺优化4、放大研究5、设备工艺研究6、合同制造7、培训反应器采用玻璃材质或无压烧结碳化硅材质,化学兼容性强,Chemtrix产品系列Labtrix Start,Labtrix S1,KiloFlow,Protrix,Plantrix为用户提供了从研发到生产阶段各种用途的流动反应合成系统。一正科技简介:作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的独家代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
新品
2021.11.25

【Purdue大学案例】Suzuki-Miyaura交叉偶联反应的高通量筛选及连续流验证
【背景介绍】自动化高通量实验(Automated high throughput experimentation,HTE)的发展,丰富了我们对化学反应范围和局限性的认识,提高了实验室的生产效率。这项技术已经影响了生物、药物发现、药物化学和催化等许多领域。自动化反应通常可以并行进行,但由于色谱分离以及定量方法相对较慢,下游分析通常是一个瓶颈。因此当其与快速质谱 (MS) 分析相结合时,可以加速反应条件的发现和优化。特别是在化学工艺开发以及(生物)药物开发中,各企业面临缩短上市时间的压力等问题便可以迎刃而解。不过,在优化有机反应条件时,特别是在可能使用固体催化剂或挥发性有机溶剂的催化反应中,自动化高通量实验(HTE)面临不少的挑战。使用HTE进行反应优化的中心目标是找到一组特定前驱体合成中最突出的反应热点条件(即反应效果比较好的实验条件)。在HTE之后,对这些热点条件进行快速验证,以建立对HTE过程确定的反应优化条件的正确性。微流控反应是在连续流动条件下验证有机反应的重要步骤,这对于催化反应必须要在更快和更环保的条件下能够实现来这一要求来说尤其如此。此外,利用电喷雾电离质谱 (ESI-MS) 对小分子有机分子的快速连续反应监测,对快速优化连续生产方法具有更大的应用前景。2018年,Purdue大学的David H. Thompson课题组以Suzuki—Miyura (S-M) 交叉偶联反应为模板,以ESI-MS为检测工具,进行了HTE反应优化的研究,采用荷兰Chemtrix 全自动微通道反应器Labtrix S1用于微流控反应的控制,相关结果发表在Chem. Eur. J. 2018, 24, 9546 – 9554【DOI : 10.1002/chem.201801165】上。图1 [ 1, 1’-联苯]-4-醇类化合物及其在一些重要的生物、医药和材料科学中的应用由于钯催化的S-M偶联形成碳-碳键是小分子合成的重要反应,对其探讨具有理论和实际的意义。近年来,Perera等人和Santanilla等人报道了高通量筛选的S-M交叉偶联反应,并证明了在不同条件下能在纳摩尔级别快速筛选反应。因此作者试图探索S-M交叉偶联反应在生物、药物和材料应用中重要的前体化合物的合成中的应用、并介绍了一种简单、有效的识别和优化S-M反应条件的方法。很多重要的联苯中间体(如图1所示)是在不需要保护官能团的情况下即通过S-M交叉偶联反应合成的。HTE所努力的重点是确定优选的反应参数,通过消除流动化学筛选过程中的失败反应条件,能够更快地优化微流体反应。图2 在S-M交叉偶联反应的高通量筛选实验中评估的底物范围S-M反应在一定的流动合成条件下已有报道,作者主要探讨了在微流体反应器中使用EtOH为溶剂时在合成重要的化合物时的应用。以4-羟基苯基硼酸和11种不同的卤代芳烃(如图2所示) 为原料,加料顺序、立体选择性、温度和浓度作为自变量,在96孔板上进行了S-M反应的自动HTE实验。以1, 8-二氮杂双环[ 5.4.0 ]十一碳-7-烯 (DBU) 为碱,XPhoPdG3为催化剂,在数百个不同的反应条件下,筛选到优化的间歇反应条件。并进一步通过HPLC/MSMS分析,利用间歇反应筛选的最佳条件,放大到小规模微流体反应中,并生产出了收率达98%的S-M偶联产物,进而验证了HTE的研究结果。 【实验过程】1. 高通量筛选实验:采用高精度Biomek FX机器人制备纳升级反应混合物,通过下游MS分析,自动筛选S-M反应条件。反应在四个96孔板中进行,其结果使用高度灵敏的三重四极质谱联用自动进样器监测,以快速测定反应产物的分布。图3 HT反应的结果与试剂加入顺序和反应混合物中底物浓度的关系以4-羟基苯基硼酸 (1) 、不同卤化芳基化合物为原料及XPtherPdG3作为钯催化剂(图2),在乙醇溶液中考察了不同卤化芳基化合物对反应效率的影响。将反应混合物(每个孔的总体积为45微升)一式两份沉积在四个96孔玻璃内衬铝阱板中,并将反应在50、100、150或200 ℃下加热1小时,然后淬灭至20 ℃,离心并稀释到384个阱板中进行MS分析。图3中的每个小方格代表一个独特的反应条件。根据反应产物峰强度报告了结果,以便能够进行简单的比较。图4 在不同温度和化学计量条件下形成的苯甲醚型联苯产品的偶合效率第一次HTE实验通过在EtOH中依次加入1 ,碱和催化剂溶液的混合物,然后加入卤代芳基底物 (2-12) 来探索S-M反应。然而不幸的是,在MS中未检测到产物峰,却观察到与水解硼酸产物相对应的峰。这些结果表明,在反应开始前的20 min内,在20 ℃下机器人试剂转移过程中,该催化剂在空载状态下遇碱而中毒。当加成顺序改变为卤化芳基和1,然后是碱和催化剂时,在所得MS中观察到产物峰,尽管产物产率并不令人满意(图3,上半部分)。这是因为在添加催化剂之前1与碱发生反应,使得没有足够的硼酸可参与催化循环。在过量的硼酸(例如,在20:1-2:1的化学计量比下)条件下,MS中生成的主要离子峰的观察进一步支持了这一假设。尽管如此,当在催化剂之后添加碱溶液时,观察到产品转化显著增加(图3,下半部分),同时MS检测到自偶合的1-1产品。作者随后利用图3中所确定的最佳条件来更广泛地筛选卤化芳基底物。实验结果表明,富电子和缺电子芳基卤化物(2-12)都能被耐受,并以中等至良好的产率生产交叉偶联的产物(图4A-D)。通过苯甲醚类的化合物之间(2-5)的比较显示,间位甲氧基(4,图4C)提供了比邻位(5,图4D)或对位(2和3,图3A和B)更好的结果,因为间位的供电子效应最小。图5 在不同温度和化学计量条件下形成的苯胺型、吡啶型联苯产品的偶合效率卤代苯胺(6-11)也能够转化为相应的联苯产品,以中等峰值强度产生了交叉偶联产物(图5A-G)。与甲氧基取代的化合物相同,间位产物17(图5C)产生的峰强度高于对位产物16(图5A和B)。当比较苯胺和苯甲醚衍生的产物时,发现苯甲醚类的产物峰强度较高,比苯胺系列的产品峰强度高13-15,16-20(图4和图5)。这是由于氨基比甲氧基取代基的电子贡献能力大,从而导致氨基苯基钯中间体的形成即在苯胺系列的催化循环的氧化加成限速步骤中不太有利。二级胺和三级胺底物的产品峰强度高于一级胺的产品峰强度(图5A-C与图5D-F),这是由于空间位阻的因素,一级胺比二级或三级胺更容易与钯催化剂配位所导致的。在吡啶衍生物中,由于吡啶环相对于苯胺系列的电子相对缺乏,观察到了较高的产物峰强度(图5G对图5A-F)。图6显示了504个独特Suzuki-Miyaura反应的产品峰强度,分别筛选每一种芳基卤化物和4-羟基苯基硼酸的反应,反应溶液中同时存在DBU和10%的 XPtherPdG3。作者采用> 50%的峰强度作为选择标准,选择了该图中的反应热点条件,用于一系列微流体反应的后续评价。图6 504个独特的S-M反应中观察到的产物峰强度2. HTE实验结果的微流体评价图7 用于S-M交叉偶联反应连续流合成的荷兰Chemtrix微通道反应器和流体处理方案从HTE实验中得到反应热点条件后,作者对每个反应的一个有利条件进行了测试,以确定对两种不同反应形式下(即批次96孔法与连续流动法)反应结果的相似程度。对于所有的微流体反应,采用1:1的4-羟基苯基硼酸和卤化芳基反应,在100或150 ℃温度下分别进行保留时间为0.5、1、3和6 min的实验评价。实验中,使用10% XPhoPdG3催化剂负载的反应在反应开始后不久就会出现堵塞的问题。通过将催化剂的负荷降低到0.5% ,可消除了这一问题。以往的工作表明,在连续流动条件下,S-M反应会更加有效,因为在较窄的反应器通道中,S-M反应具有更好的传质传热和更高的混合效率。由于所有试剂都在同一混合区内接触,因此添加顺序在微流体合成中并不重要(图7)。TLC和MS证实了预期产物的形成。如图8中的数据所示,微流体连续反应的结果与高通量筛选的结果基本相当。图8 用200 mmol底物负载和1:1 4-羟基苯硼酸:卤化芳基化学计量比在相似条件下进行S-M反应的微流体反应和高通量筛选结果的比较 图8中的数据表明,S-M反应允许芳基卤化物底物中的电子供体以提供具有不同效率的交叉耦合产物。在两次实验中中,间位苯甲醚的联苯产物的产物转化率要高于邻位。但在连续流动过程中,对位取代的产物也显示出较高的产物峰强度。类似地,大体积叔胺和吡啶取代底物在两种形式实验中都产生了更高的产物峰强度。作者还研究了在HTE实验的负结果的条件,并在连续流动条件下重新评估了这些反应条件(图9)。实验结果表明,即使延长时间,也没有得到所需要的产物。图9 S-M反应的微流体和本体筛选结果的比较,均得到了负结果接下来,作者重点测试了用产物离子强度来衡量反应进程的有效性。由于苯甲醚和吡啶取代的反应产物在高通量筛选和微流控反应中均表现出较高的峰强度,因此选择其作为HPLC/MS-MS的评价指标。观察到所有苯甲醚(2-5)和吡啶12起始原料的总的产品收率随时间的增加趋势(图10)。从筛选结果来看,14和21的收率都很高。在大多数情况下,峰值强度和定量产品产量之间有很好的一致性。由于o-MeO取代的底物需要更多的能量来驱动反应,因此15的产率较低。 图10 S-M微通道反应收率与使用HPLC/MS-MS的峰强度的比较【实验过程】1、 这项研究使用机器人HTE技术在96孔阵列中执行反应,随后使用自动取样器进行快速MS分析。对钯催化的S-M交叉偶联反应进行了筛选,得到了反应热点条件图,为在连续流动中的下游放大提供了条件。2、 用4-羟基苯基硼酸和11种不同芳基卤化物重复进行了648个独特的实验研究,结果表明反应的产率与MS峰强度有关。3、 采用荷兰Chemtrix全自动微通道反应器Labtrix S1, 在微流体条件下对HTE的一些成功反应进行了比较,这些实验证实了HTE所确定的阳性条件在连续流中是真正的阳性结果。此外,在微流体条件下,即使在较高的温度下长时间停留,该方法所确定的负反应条件也会产生负结果。HPLC/MS-MS定量分析为HTE反应筛选和微流体反应提供了良好的证据。4、 这种HTE和微流体验证方法同样适用于其他催化和非催化反应,用来指导反应优化工作。这种方法也可用于确定挑战性底物的最佳条件,或发现生产生物活性分子的新途径。下一步实验将需要沿着这些用途进行进一步的实验,以评估这种相关性的稳健程度。 原文:Chem. Eur. J. 2018, 24, 9546 – 9554【DOI : 10.1002/chem.201801165】
厂商
2020.03.13

【加拿大Apotex奥贝泰克制药公司案例】连续流还原脱氧工艺生产维拉唑酮合成中的ω-氯酮中间体
【背景介绍】 盐酸维拉唑酮(图1)是用于治疗重度抑郁症的3-烷基吲哚药物。1的釜式生产方法是已知的,其中一种方法是中间体4a-c与中间体3a/b反应而得(如图2),而3a/b是由化合物(例如2a/b)的还原性脱氧获得的。例如,可以在硼氢化钠和三氯化铁为还原剂的条件下,将将3-(4-氯丁酰基)-1H-吲哚-5-腈(2b)还原为3-(4-氯丁基)-1H-吲哚-5-腈(3b)。而在具体的实施过程中,为了控制还原剂加料过程中的热量,因此在低温下进行,导致了难以去除的杂质(5)的积累。去除这种杂质需要额外的处理,便可能导致3b的产率的降低。连续流对反应放热有着出色的控制,进而可以使反应在较高的温度下运行并同时可以减少反应时间,抑制了副反应和杂质的形成。正是由于这些原因,加拿大Apotex奥贝泰克制药公司的Prabhudas Bodhuri等人,以连续流模式探索2b的还原性脱氧反应,力求在抑制杂质形成的同时,实现更高的3b收率和纯度。相关结果发表在Org. Process Res. Dev.期刊上。图1 盐酸维拉唑酮的结构图2 商业合成1的路线及副反应的产生原理 流动化学工艺在制药业和学术界的应用越来越广。化学反应可从流动工艺中受益,即减少杂质,提高产率和选择性并允许使用有害化学物质(否则将作为可能的试剂丢弃)来进行化学反应。困难在于如何增强化学反应以适应连续流动所需的条件,主要的问题是避免使用致密的固体试剂,而这会导致连续流动设备(例如推流式反应器)的泵送管线出现堵塞问题。 【实验过程】 分批过程中2b的还原性脱氧涉及将硼氢化钠固体分批添加到2b和三氯化铁在四氢呋喃中的浆液中(图2). 随着氢气的产生,反应混合物在反应过程中保持为淤浆状态。由于反应混合物的多相性质,作者决定将英国AM公司Coflore ACR连续多级搅拌反应器(CSTR)设置用于作者的研究,以避免上述机械问题。这款设备由一个反应块组成,该反应块包含通过小通道串联连接的10个反应池(图3)。由于每个单元中都有机械搅拌器而产生的浆液和悬浮液,并具有多种侧端口,可用于添加试剂,热电偶和其他过程分析技术(PAT)。可通过改变每个单元中使用的搅拌器的尺寸来改变反应器模块的内部体积。这样可以使初始池中的体积更小,以实现充分混合、传热和为放大做准备。图3英国AM公司Coflore ACR连续多级搅拌反应器(CSTR)的结构 作者致力于应用连续流化学法生产中间体3b;在三氯化铁(FeCl3)存在下在四氢呋喃中用硼氢化钠(NaBH4)对2b进行还原性脱氧反应得到3b。还原性脱氧步骤在实验室中进行了优化,并可扩大到50-70 kg的规模。观察到在最初的按比例放大批次期间,该杂质的含量随反应时间的增加而增加(图4). 用于生产的初始过程如下:在批次1中,1 h内将NaBH4固体在0-5°C下分8次加入2b和FeCl3在THF中的浆液中,然后在2小时内升温至35-40°C,再保持温度3小时以完成反应。这样虽然只产生了少量的杂质5,但是仍然需要纯化以除去其他二聚体杂质,从而导致总产物收率大约为20%。换用另一种方法,在批次2中,温度在0-5℃下保持6小时以完成反应,在此过程中,粗产物的收率略高,但杂质5的含量更高。在批次3中,反应时间在0-5°C下延长至11 h以完成反应,这使得粗品收率和杂质5水平均大量提高。由于大量的杂质5,需要进行多次纯化才能达到既定的规格限制,并且总产率仅为20%。从批次1的数据中,作者看到较高的温度使杂质5的含量变小。这可能表明中间体4在较低的温度下具有更长的存在时间,从而使其也可以与产物反应生成杂质5。作者设想将连续流技术应用到该过程中,以更快的反应时间将反应温度提高到40°C(与之相比,在间歇模式下低于40°C),从而提高了总收率并减少了所有杂质的数量。在釜式模式下,由于与在硼氢化钠添加过程中控制放热有关的安全问题,因此无法在40°C下进行反应。 还原性脱氧过程中产生的反应混合物由三个物理相组成,分别是固体NaBH4和起始原料2b,作为液体溶剂的THF和释放的氢气。搅拌池反应器CSTR装置非常适合研究这种类型的过程,因为它可以处理多相反应混合物,这弥补了不适合这些类型过程的活塞流反应器的缺点。在以连续流程研究此过程之前,必须在实验室规模下对当前过程进行一些修改。首先,将反应温度升高至40℃,在该温度下还原反应在30分钟内完成,这说明了反应在40oC进行的可行性。其次,还必须解决向反应中添加硼氢化钠的问题。硼氢化钠是在THF中的致密的不溶固体,无法有效泵送以用于连续流工艺中。但是,硼氢化钠可溶于二甘醇二甲醚中,作者的最初工作是在该过程中使用硼氢化钠的二甘醇二甲醚溶液。作者很快发现,硼氢化钠在二甘醇二甲醚中的溶解度对温度非常敏感(如图5所示)。使用饱和的硼氢化钠溶液时,即使很小的温度变化也会导致NaBH4沉淀而导致溶液管线堵塞。为了克服这个难题,作者将NaBH4溶剂改为可在较宽的温度范围内可提供更均匀的NaBH4溶解度的四甘醇二甲醚,并且相对于二甘醇二甲醚在环境温度下总体溶解度更高。图4 早期的实验室放大数据图5 硼氢化钠在二甘醇二甲醚和四甘醇二甲醚中的溶解度-温度曲线 作者的初始实验条件如下:将1.1当量的三氯化铁在THF中的溶液泵入CSTR 1,将酮2b在THF中的浆液泵入CSTR 1,并在40°C的反应温度和30分钟的停留时间下将1.2当量的NaBH4在四甘醇二甲醚溶液中泵入CSTR 2。(如图6所示). 反应结果中杂质5的量降至最低,但使用柠檬酸淬灭后仍保留约20%的中间体4。将该反应混合物取样,然后淬灭并用少量NaBH4的四甘醇二甲醚溶液的处理。这使得中间体4完全转化为3b(图7).图6 作者的初始实验流程图图7 中间体4转化的HPLC数据,可看出第二次加入NaBH4时已经完全转化 由于需要额外的NaBH4来完全消耗中间体4,因此在CSTR 6处将设置第二个NaBH4的添加端口,并装入0.4当量的NaBH4。新的工艺设置将1.1当量的NaBH4添加到CSTR 2中,并将0.4当量的NaBH4添加到CSTR 6中,从而可以以34-45%的产率分离产物3b,HPLC纯度为99.5%,杂质5含量为0.09%。 溶液中使用NaBH4溶液进行连续流动过程能够降低杂质含量并提供高纯度3b,但是由于约23%的分离产率较低,产品损失进入母液。为了提高收率,主要通过消除对四甘醇二甲醚作为助溶剂的使用,探索了其他还原剂以促进更简单的后处理和分离。发现在三氯化铁存在下,硼烷四氢呋喃(BH3•THF)配合物是极好的还原剂。稍作优化后,作者发现向CSTR 1中加入1当量的三氯化铁,然后向CSTR 2中加入1当量的BH3•THF,CSTR 6中加入0.2当量的BH3•THF在20-25°C的温度下将反应,并将保留时间设置为5分钟。这些条件使得3b的分离收率为66.5%,HPLC总纯度为99.6%,杂质5含量为0.09 %. 收率提高21%是由于使用BH3•THF后进入母液的产物量减少使得损失减少。与5 g规模的釜式反应进行对比,由于在添加BH3•THF的过程中观察到很剧烈的放热现象,该反应必须在0-5°C下进行,同时需要分6批添加BH3•THF。在每次添加BH3•THF的过程中,内部温度都会升高5-6°C,并且在添加最后一部分后不久,反应完成。后处理和分离后,获得64%的3b,HPLC纯度为99.39%,杂质5含量为0.17%。而3b的规定的标准为> 99.50%的HPLC纯度和 作者进一步指出,连续流动处理的技术挑战之一是由于硼烷试剂与吲哚环的酸性质子反应以及在柠檬酸水溶液中过量硼烷的淬灭而在搅拌池中产生的氢气积聚。根据上述氢气积累的可能问题,为了评估将该工艺规模扩大到千公斤或中试工厂的可行性,作者研究了由夹套反应器组成的CSTR装置在该工艺中的使用。使用两个20 mL夹套反应器和一个150 mL夹套反应器构建一个带有夹套反应器的CSTR装置,工作容积为30 mL。每个CSTR连接到氮气管线,以使产生的氢气逸出。作者的初始设置是在20°C下将三种试剂同时添加到CSTR 1中。但是,这导致内部温度从20°C升高到36°C。为了消散试剂混合过程中高放热还原反应产生的大量热量,在进入CSTR 1之前,装一个磁力搅拌器,并将冷却一段反应管道,将其保持在20oC。使用夹套冷却系统将三个CSTR反应器维持在20-25°C左右。并将第二部分BH3•THF添加到CSTR 2中,将反应混合物离开CSTR 3后,用柠檬酸水溶液淬灭(图8). 使用该设置,作者以71%收率得到3b,HPLC总纯度为99.6 a%,杂质5含量为0.06 a%。图8 2b还原的扩大实验:由夹套反应器组成的CSTR装置示意图 通过对现有釜式方法、NaBH4连续流动法和BH3·THF连续流动法三种不同工艺的比较,说明了连续流动工艺在该化工过程中的实际应用价值。从间歇过程到连续流动过程的变化有几个有利的方面,即过程安全性、单元操作次数、过程质量强度(PMI)、和总分离收率(图9)等等。与使用NaBH4还原剂的间歇过程相比,在连续流动过程中,单元操作次数和PMI值均减少,并且产品收率进一步提高,由于使用了更清洁的化学转化,从而消除了CF过程中的净化步骤。在BH3·THF连续流动过程中,产率进一步提高,单元操作次数进一步减少。图9 三种不同工艺的对比 【结论】1. 连续流动过程对维拉唑酮中间体3b合成的优势是显而易见的。通过在英国AM公司Coflore ACR连续多级搅拌反应器中进行反应可以成功克服分批反应中控制放热的局限性,该装置也能够处理异质混合物。2. 连续流动过程减少了关键的二聚杂质5的产生,从而提高了产物3b的产率。结果表明,利用Coflore ACR反应器仪器进行连续流过程研究,并对反应条件进行一些修改(例如,使用四甘醇二甲醚中的NaBH4作为还原剂),可以提高分离出的3b的收率和纯度。甚至可以达到不再需要纯化过程的程度。3. 使用BH3•THF作为还原剂,可进一步提高了收率,得到无需纯化的高纯度产品。使用三夹套反应器系统将BH3•THF工艺扩展到CSTR设置,与现有的釜式工艺相比,该工艺生产3b收率更好、纯度更好。 【公司简介】 作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。 公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。 公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及优质的技术服务方案。
厂商
2020.02.28
【印度理工大学案例】固定化Rhizopus microsporus IBBL-2海藻酸钙微球在Spinchem旋转床反应器中生产L-天冬酰胺酶
【背景介绍】L-天冬酰胺酶(酶代码EC3.5.1.1,ASNase)是一种催化天冬酰胺脱氨为天冬氨酸和氨的氨基水解酶。这种酶广泛应用于成人、儿童急性淋巴细胞白血病和儿童非霍奇金淋巴瘤的治疗。L-天冬酰胺酶也用于食品工业,以防止食品在油炸或烘烤过程中形成丙烯酰胺。另外,它也可以作为一种生物传感器的应用,用于检测所有化疗期间的天冬酰胺水平。对于正常细胞来说,L-天冬酰胺是一种非必需氨基酸,它可以通过天冬酰胺合成酶以必要的量合成。正是这种酶将体内的谷氨酰胺转化为细胞生长所需的天冬酰胺。相反,白血病细胞缺乏天冬酰胺合成酶,而其恶性生长则需要大量的L-天冬酰胺,因此依赖于血浆中L-天冬酰胺的外源供应。L-天冬酰胺酶可通过水解肿瘤细胞所需的L-天冬酰胺,从而使其饥饿并杀死白血病细胞。天冬酰胺缺乏导致蛋白质合成不足,DNA和RNA合成延迟,从而损害细胞功能,导致癌细胞死亡;由于这种对白血病细胞的选择性作用,L-天冬酰胺酶被认为是治疗ALL的里程碑。L-天冬酰胺酶可以用不同的细菌、真菌、酵母和放线菌等组织合成。在目前的情况下,临床上有三种L-天冬酰胺酶制剂,即来自大肠杆菌的天冬酰胺酶、菊欧文氏杆菌和聚乙二醇化大肠杆菌。临床配方的L-天冬酰胺酶对谷氨酰胺、尿素有亲和力,而且酶的半衰期降低了;因此,患者表现出特殊的过敏反应。因此,寻找特异性高、过敏反应低的L-天冬酰胺酶是研究者面临的一个特殊挑战。为了去除这些副产品,必须对产品进行高纯度的纯化。目前正在进行广泛的研究,以寻找细菌种类的有效替代物,并生产不含谷氨酰胺酶和尿素酶的L-天冬酰胺酶。在任何工业中,酶生产的主要关注点是其低稳定性和低活性的特点;因此,固定化技术的使用有助于提高任何工艺的生产率。通常,根霉可以在以葡萄糖为碳源的简单培养基中生长,但由于菌丝生长导致培养基粘度增加,因此用丝状真菌发酵较为复杂。全细胞固定化是一种将细胞限制在空间内而不丧失其生物活性的技术。细胞可以用不同的物理或化学方法支撑在固体基质结构上;最常用的技术是夹带整个细胞,这种技术被称为生物包膜或微胶囊化。Tay和Yang报道了在旋转纤维床生物反应器中固定化米根霉细胞用于乳酸生产,可以实现几乎无细胞发酵,并且比悬浮真菌细胞提供更高的产量。这种技术的使用还减少了酶的提取和纯化所需的时间,因为被固定化的整个细胞具有更好的反应能力。使用何种固定化技术的决定取决于所用细胞的性质、其与固体载体的反应性以及所生产材料的类型。印度理工大学的Devarai等人通过用海藻酸钙(Ca-alginate)珠包埋全细胞根霉小孢子IBBL-2生产L-天冬酰胺酶。研究了海藻酸钠溶液浓度、小球大小等因素对酶活性的影响。该实验结果发表不久前发表在3 Biotech期刊上。(https://doi.org/10.1007/s13205-019-1883-5)Spinchem旋转床反应器(RBR)是一种填充有固相的填料床,在其研究案例中,固相是含有微生物的固定化珠。该反应器基于离心加速度工作,这有助于增强在反应器中发生的混合和传质操作。使用该反应器进行过程强化,不仅降低了过程的经济性,而且增加了总产量。在RBR中,培养基通过包装,在固定化材料内进行培养基与微生物之间的发酵过程,酶作为输出被释放。由于转子中的小球很致密,与自由流动的液体相比,整体应力降低,这有助于提高酶的活性。同时,作者也采用200mL和1L的RBR进行了放大研究,以证明该工艺的有效性。实验的装置如图1所示。图1 实验装置和程序示意图【实验过程】pH值的影响采用单因素一次法对各因素的工艺效率进行了分析。为了使微生物正常生长,必须确定每一个参数;任何低于或高于最佳值的值都会对微生物的生长产生不利影响。在30℃的温度下生产l-天冬酰胺酶。IBBL2在MCD培养基上生长,培养基的pH值在4-8之间。pH值对L-天冬酰胺酶的影响如图2A所示,小根霉菌IBBL-2在微酸性pH下表现出更好的活性,活性曲线表明酶释放或微生物生长在实验的第二天和第三天之间迅速上升,其最大活性出现在第三天。结果表明,在pH为6时,酶活力最高,活性为11.69U•mL-1。然而,在低pH(4和5)和高pH范围(8-9)时,天冬酰胺酶活性都很低。温度的影响培养温度对发酵过程中微生物的生长和酶的产生有着深刻的影响。用预先优化的pH值在15-50℃范围内研究了温度对L-天冬酰胺酶活性的影响。L-天冬酰胺酶活性和不同温度下的比活性如图2b所示。当前的研究表明,小根霉IBBL-2生长的最适温度为30℃,在此温度下最大活性为12.2 U•mL-1,比活力为19.29 U•mg-1。在低温和35℃以上的高温下均观察到L-天冬酰胺酶活性的下降。初始孢子浓度的影响作为微生物生长接种物,所添加的初始细胞数是另一个需要研究的重要因素。初始值越低,微生物的生长就越不正常,而初始值越大,微生物的生长就越拥挤,导致初始生长所需的养分缺乏。初始孢子浓度对小孢子菌IBBL-2产生L-天冬酰胺酶的影响如图3A所示,目前的研究表明,在每毫升4×106个细胞浓度下,获得的最大L-天冬酰胺酶活性为12.02 U•mL-1。图2微孢子根霉IBBL-2在不同pH(a)和温度(b)条件下的液体发酵l-天冬酰胺酶活性和比活力 图3利用微孢子根霉IBBL-2分别改变微生物细胞初始浓度(a)和金属离子类型(b)的液体发酵中的l-天冬酰胺酶活性和比活性 金属离子效应影响微生物生长的另一个因素是金属离子,根据其对微生物生长的影响,金属离子可以促进或减少微生物的生长,也可以作为几种生物合成酶的共同因子。从实验结果看,Cu2+对生长有积极的影响,其最大活性为13.92 U•mL-1,如图3b所示。在测试的金属离子中,当在MCD培养基中添加Na+金属离子时,酶活性最低。 碳氮源效应使用各种碳源(葡萄糖、果糖、淀粉、乳糖和蔗糖)对L-天冬酰胺酶生产影响如图4a所示。微生物的生长受正在供应的营养源的控制。碳源是任何微生物发育的主要因素。以葡萄糖为碳源,小孢子根霉菌IbBL-2的最大活性为14.43μU•mL-1。从图4a可以确定,小根霉IBBL-2与葡萄糖的配伍性最好,以蔗糖或果糖为唯一碳源时,L-天冬酰胺酶活性下降。通过添加氮源(天冬酰胺、谷氨酰胺、蛋白胨、酵母抽提物、硝酸钠和硫酸铵)研究了氮化合物对微孢子虫IBBL2产l-天冬酰胺酶的影响。氮源为生长所需蛋白质的生产提供物质。如图4b所示,微孢子根霉IBBL-2菌株对L-天冬酰胺具有更高的亲和力(L-天冬酰胺酶活性14.18 U mL-1)。酵母抽提物也能够支持L-天冬酰胺酶的大量生产。以硝酸钠为氮源时,L-天冬酰胺酶活性最低。作者又通过Taguchi OA 方法进一步优化了实验条件,当固定化海藻酸钠浓度为3%,细胞珠体大小为2mm时,反应效果最好。图4 利用微孢子根霉IBBL-2分别改变微生物碳源(a)和氮源(b)的液体发酵中的l-天冬酰胺酶活性和比活性 大规模生产L-天冬酰胺酶采用优化的培养基组成和最佳条件(pH6.0,30°C,接种量25ml,3%的固定化材料浓度,2mm大小的珠体),在锥形瓶和固态托盘生物反应器中进行了L-天冬酰胺酶的实验室规模生产。在本研究中,制备了200ml和1l容量的合成培养基,并用RBR对固定化海藻酸钙珠进行了反应。由所得结果可知,活性值与烧瓶研究所得值相近。图5所示的实验室规模RBR研究获得的活性为200mL 实验组为20.21 U m L-1 、1L实验组为19.13 U m L-1。固定化酶活性的增加归因于细胞的稳定性增加。这导致了介质的稳定输入和产品的输出;同时,由于剪切力是细胞中可能发生的损伤的原因。而在RBR介质的流动中,细胞不受任何剪切力的影响,进而受到保护。图5 半中试扩大培养中,酶活(a)和比活性(b)在200ml和1L的比较 【实验结论】1. 在固定温度下,以葡萄糖为碳源,L-天冬酰胺为氮源,以Cu为附加金属离子,在30℃、pH为6、初始微生物浓度为4×106个细胞每mL的条件下使用3%(W/V)浓度下制备的粒径为2 mm珠体进行培养,小孢子根霉菌iBL-2真菌的最大活性为17.68 U mL-1。比未固定化的活性增加了1.4倍。2. 且作者通过放大研究表明,生产酶活性在200mL级旋转床反应器和1L级旋转床反应器的放大过程中没有损失。3. 小根霉IbBL-2能产生不含谷氨酰胺酶和脲酶的L-天冬酰胺酶,从而取代了现有的利用细菌种类的生产。它在固定化技术的应用中也显示出良好的稳定性。 文献原文:https://doi.org/10.1007/s13205-019-1883-5l?Asparaginase production in rotating bed reactor from Rhizopus microsporus IBBL?2 using immobilized Ca?alginate beads 【公司简介】作为荷兰Chemtrix微通道反应器(适合液液气液快速反应),英国AM连续多级搅拌反应器(适合气液固多相慢反应),瑞典SpinChem旋转床反应器(酶催化,固定化酶,催化剂需要回收的反应),澳大利亚CSIRO催化剂固定化连续反应器(适合催化剂固定的连续流反应),比利时Creaflow光催化反应器(气液固光催化反应),英国C-Tech电化学连续反应器,英国Nitech连续结晶器,德国CINC连续萃取分离器,英国AWL连续过滤器在中国区的代理商和技术服务商,深圳市一正科技有限公司为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及优质的技术服务方案。
厂商
2020.02.14
采用荷兰Chemtrix微通道反应器快速按需合成抗癌药物洛莫司汀
【普渡大学2019 OPRD案例】采用荷兰Chemtrix微通道反应器快速按需合成抗癌药物洛莫司汀【背景介绍】洛莫司汀(Lomustine),是一类被广泛使用的高亲脂性烷基化抗癌药物,通过在体内产生氯乙基碳离子和碳酰化中间体,这些亲电化合物攻击DNA上的亲核部位,形成烷基化产物,从而发挥药效作用的。其他类别的抗癌药物,比如丝裂霉素C、链霉素、博来霉素和蒽环类,需要生物激活作用(bioactivation)来与细胞靶点反应,而洛莫司汀不需要预激活。不同于作用于鸟嘌呤的N7烷基化试剂,洛莫司汀则在O6形成氯乙基化加合物,进而导致链间DNA的交联。如果DNA不进行修复,这种交联会在DNA复制过程中导致双链断裂,最终细胞凋亡导致细胞死亡。洛莫司汀作为一种可口服抗肿瘤药,每6周服用一次。它最早是在20世纪60年代后期进行的临床试验评估,并于1976年获得FDA的批准,用于治疗原发性和转移性脑肿瘤以及霍奇金淋巴瘤。百时美施贵宝最初以CeeNu品牌持有该药物的专利。2014年,FDA批准Next Source Biotechnology公司被以Gleostine的商标对洛莫司汀进行重新包装。最近,Gleostine的平均批发价格出现了大幅上涨,其原因是处理致癌化合物所产生的监管风险 。因此,这促使我们努力开发一种通过连续流动技术合成洛莫司汀的方法。流动化学技术最近在工业界和学术实验室中都进行了广泛的探索,被证实是一种有效的合成技术。与传统的釜式合成工艺相比,由于连续流技术可以快速混合和精确控制反应参数,进而更好地控制反应条件和选择性,如温度、化学计量、压力和保留时间。强化的传热和传质能力也提供了更安全的操作条件。连续流合成的工艺强化可以有助于提高化学反应效率,缩短反应时间,减小反应规模,提高产品的质量和一致性。因此,原料药的连续流合成技术在最近几年变得极具吸引力。然而,由于工作条件、溶剂切换和流速差异等因素的限制,通过连续流技术有效地执行多步反应仍然是一个挑战。此外,连续流方法的反应条件的优化和分析技术仍需要大量人力物力的投入。【连续流方法】Purdue大学的David H. Thompson课题组近日报道了洛莫司汀通过连续流合成的新工艺方法,并使用解吸电喷雾电离质谱(DESI-MS)对其进行分析检测指导工艺的优化方案。最终工艺路线是两个反应串联在一起,不分离中间体,具备反应器装置简易、原料廉价等特点因此可以降低生产成本(反应流程如图1所示)。相关结果发表在Org. Process Res. Dev.2019,23,3,334-341(DOI:10.1021/acs.oprd.8b00387)杂志上。图1 使用连续流技术两步串联法合成洛莫司汀的工艺路线,其中4 = NaNO2/HCO2H ,5 = tBuONO洛莫司汀合成的第一步在室温下是快反应。在连续流中对该反应的温度、溶剂和投料比进行优化,进而确定获得最大产品收率的条件。检测反应使用TLC和MS,其中MS使用正离子模式下三重四极质谱仪,以快速探测出每种反应条件下的全质谱和产物离子分布。为了揭示第一步的最佳条件,作者设计了一种级联方法。运用荷兰Chemtrix微通道反应器,选用 3225 SOR静态混合区芯片,泵送环己胺(1),1-氯-2-异氰酸乙酯-乙烷(2)和三乙胺(TEA)溶液,自动进样器自动收集小瓶中的产品(如图2所示)。蒸发反应混合物,用冷Et2O洗涤白色固体并真空干燥,然后用TLC、MS、MS/MS和NMR(1H和13C)分析。图2 合成中间体 3的Chemtrix微反应器 glass reactor chip (SOR 3225). A = 1 in THF; B = 2 in THF, C = TEA in THF.作者对该步反应使用的保留时间(τ)、反应温度(T)、1-氯-2-异氰酸乙酯/环己胺比等条件进行探讨。如图3所示,在较长的保留时间下,EtOH为溶剂的产品产量急剧下降(Entry1-3),原因是因为1-氯-2-异氰酸乙酯会被溶剂醇解,尽管3在ACN中的收率很高,但它在该溶剂中的溶解度很低,导致严重堵塞。对于甲苯和Et2O,即使在低浓度下也发现了类似的堵塞问题(Entry7-8)。而在溶剂THF中,产率在在50 oC下30 s达到最大值(Entry9-14),并且由于高温会导致产物分解,温度升高,产率会下降(Entry13-14)。提高2的当量可以提高产率(Entry15-21),而延长保留时间,也可能会导致产品分解(Entry21)。最终,作者确立了最佳的反应条件:τ = 60 s, T = 50 °C, 1.4 当量的1-氯-2-异氰酸乙酯。在该条件下,反应的最高产率可达到92%(Entry 20)。图3 合成中间体3的反应条件筛选对于第二步的合成,作者使用DESI-MS来评估溶剂、浓度和硝化试剂类型对洛莫司汀产率的影响。DESI-MS最初应用于完整样本的表面分析,如癌症诊断用生物组织或药物检测指纹图谱。在最近,DESI-MS方法已用于化学反应分析。该方法主要目的是在受限的体积内产生反应加速现象,比如在MS喷雾电离过程的微液滴中,通过这种技术可以达到5ms的分析速度,每小时接近一万个反应点。作者使用了这项技术来研究两种亚硝化方法,首先,评价了亚硝酸钠、甲酸作为亚硝化试剂对中间体3向洛莫司汀转化的效率。然后将该转化率与亚硝酸叔丁酯(TBN,5)作为亚硝化试剂进行比较。用DESI-MS研究了NaNO2/HCO2H体系不同反浓度和溶剂条件下的转化效果。在探究中,洛莫司汀的分子离子峰m/z= 234未被观察到。对市售洛莫司汀样品的分析两者产生了非常相似的质谱,这表明电离过程中该分子很容易发生碎裂。通过三重四极质谱与电喷雾电离,以及离子阱质谱联用DESI,质谱中得到了一个稳定的固体碎片离子在m/z 169。该反应产生的洛莫司汀碎片与市售的洛莫司汀相匹配这一事实进一步证实了这一点。THF/H2O中所有NaNO2浓度的反应比ACN/H2O的反应效果更好(图4a)。此外,与THF/H2O反应相比,来自3的碎片在ACN/H2O反应中最为丰富,表明起始物质在ACN/H2O中的转化相对缓慢。接下来,将8种不同溶剂(EtOH、MeOH、DCM、ACN、甲苯、DMSO、THF和EtOAC)进行结果比较优化连续流合成。所有八种溶剂都使用4作为亚硝化试剂并得到了类似的结果(图4b)。当使用5作为硝化试剂时,在除THF和EtOAc以外的所有溶剂中都检测到洛莫司汀(图4c)。图4 DESI-MS 样品盘显示了在合成洛莫司汀的条件筛选中是否存寻找的离子峰,蓝色点表示m/z169出现了(成功反应),红色点则表示没有出现(不成功反应) (A) NaNO2作为硝化试剂时的浓度筛选. (B) NaNO2作为硝化试剂时的溶剂筛选(C) tBuONO 作为硝化试剂时的溶剂筛选接下来,作者利用DESI-MS高通量实验中得到的反应条件来优化连续流合成,并检查由DESI-MS确定的不成功反应在流量条件下是否会产生负迁移。从DESI-MS实验中可知,THF是使用4进行亚硝化反应的最佳溶剂。由于亚硝化试剂是吸湿的并且容易氧化成硝酸盐,反应常常会使用过量的4,进而将3最大限度地转化为洛莫司汀。总的来说,DESI-MS实验在投料比、试剂和溶剂方面与流动反应条件的结果非常一致。在连续流反应中筛选洛莫司汀的转化率(图5)。作者使用TLC和MS检测反应。最初,在0°C、保留时间为30s的条件下。令人欣慰的是,在这么短的反应时间内获得了洛莫司汀,虽然转化率很低(图6,Entry1)。适当提高保留时间可以提高转化;然而,较长的保留时间似乎增加了对洛莫司汀的分解(Entry2-6)。同时,作者将温度保持在0°C,以避免NaNO2分解成高温下产生的重氮盐。图5 合成洛莫司汀的微反应器,在 Chemtrix 3225 SOR glass reactor chip中使用NaNO2 /HCOOH(4)作为硝化试剂,A = 3 in THF; B = HCO2H (纯), C = NaNO2 in MeOH/H2O (4:1).接下来,作者对洛莫司汀的不同分离纯化方法进行了评价。首先,用Et2O(3次)萃取产物,以利用3在该溶剂中的低溶解度(Method 1)。不幸的是,薄层色谱分析显示有机层和水层中都存在3。只好将有机萃取物经无水Na2SO4干燥并在真空中浓缩以产生黄色油,该油在Et2O中重新溶解,在冰浴中冷却以从混合物中沉淀3。对滤液的NMR分析显示,仍有22%的3残留在分离的洛莫司汀产品中。接下来,作者换用ACN(Method2)重结晶来纯化产物。虽然得到了非常纯的洛莫司汀,但使用这种方法的回收率很低。最后,作者发现使用石油醚热过滤可去除不溶性杂质3(Method3)。干燥后滤液浓缩得到纯的洛莫司汀,通过NMR分析检测不到3。采用此方法,在0°C,保留时间为5分钟,3当量4的反应条件下,以74%的分离收率得到纯的洛莫司汀。图6 使用4作为硝化试剂时,不同反应条件下洛莫司汀的分离收率当使用5作为亚硝化剂时,作者也在保留时间、温度和溶剂方面对反应进行了优化(反应装置如图7所示)。结果表明,在保留时间(8分钟)、降低温度(25°C)和增加5的比率(3当量)的条件下,通过方法3纯化后,洛莫司汀有了进一步的提高(91%分离收率)。图7 合成洛莫司汀的微反应器,在 荷兰Chemtrix 3225 SOR glass reactor chip中使用tBuONO(5)作为硝化试剂,A = 3 in ACN/ EtOH (3.7:1); B = 5 in ACN.下一步研究了连续流装置下的两步串联反应,作者使用荷兰Chemtrix反应器来观察碳酰化和亚硝化反应。然而,在两个步骤之间需要萃取,以去除第一反应中存在的三乙胺,避免在第二步骤中竞争性地消耗亚硝化试剂。作者通过在亚硝化步骤之前使用市售Zaiput液-液萃取器去除碱,并通过使用FEP管式反应器重新优化合成来实现这一目标(图8)。开始时,最佳反应条件为:第一步在50°C下,保留时间1分钟,第二步在0°C下,保留时间5分钟,得到43 mg(总收率51.8%)的纯洛莫司汀(图9,Entry 1)。在使用4作为亚硝化试剂的条件下,通过改变第一步或第二步的保留时间来提高FEP管式反应器中洛莫司汀产率是不能成功的(Entry 1-4)。图8 利用4做为硝化试剂,两步串联法合成洛莫司汀的实验装置而在随后的实验中结果表明,5比4是一种更好的合成洛莫司汀的亚硝化试剂。最终的优化条件包括第一步在50℃下的1分钟反应时间和第二步在25℃下的8分钟反应时间(如图10所示)。由于从第一步提取的少量三乙胺对TBN活性产生了影响,降低所用三乙胺的量使得洛莫司汀产量增加(表4,Entry 7-9),通过该反应装置制得110 mg的洛莫司汀(表4,条目9),总收率为63%。通过这种串联反应路线制得的洛莫司汀的经TLC、NMR(1H和13C)、MS和MS/MS表征与市售的洛莫司汀样品的测定值相同。图9 串联反应合成洛莫司汀的条件筛选采用高效液相色谱-质谱联用(HPLC-MS)对合成的洛莫司汀进行纯度鉴定,并将该物质的总离子色谱(TIC)值与市售的洛莫司汀标准品进行比较。TIC与商业标准品完全重叠,没有出现任何新的副产品。图10 利用5做为硝化试剂,两步串联法合成洛莫司汀的实验装置【实验结论】1.作者利用DESI-MS开发了一种快速连续合成洛莫司汀的方法,指导了二步整体转化中第二步反应条件的优化。2.该工艺采用9分钟的总保留时间,以63%的总分离收率生产纯品洛莫司汀,与釜式反应相比,无论从收率和效率方面都有了很大的提高。3.亚硝酸叔丁酯(5)是一种温和而高效的亚硝化试剂,并且可以通过简单的提取、过滤和洗涤来分离纯洛莫司汀。这是一个更快更绿色的合成工艺,该工艺可以显著缩短反应时间,减少废物产生,避免任何色谱分离步骤。使用这种方法,每小时可以产生110 mg的洛莫司汀。同时作者也在对洛莫司汀放大生产和在线再结晶进行探讨。ReferenceOrg. Process Res. Dev.2019,23,3,334-341公司简介:深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司(连续多级搅拌反应器、催化加氢系统)、英国NiTech公司(连续结晶仪、连续合成仪)在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
厂商
2019.07.22
吲哚-3羧酸酯及其类植物生长素衍生物的流动合成
【英国杜伦大学案例】吲哚-3羧酸酯及其类植物生长素衍生物的流动合成吲哚类化合物是色氨酸(1)、相关的神经递质血清素(2)以及许多复杂的生物碱天然物质和药物中常见的最重要的生物活性杂环结构之一。吲哚类植物激素在促进和调节植物生长发育方面也发挥着重要作用。的确,80%的植物自身合成的生长激素含有吲哚结构(图1,结构3-7)。由于它们的调节活动,这些结构已成为研究促进植物生长以及目标植物的生长抑制作用产生新型农用除草剂的主要目标。最近的一项合作研究了合成的吲哚-3-乙酸类似物在阔叶植物(双子叶植物)中的吸收及其分布,需要大量制备结构8(图1),以进行大规模的田间试验。此外,由于气候模式和环境问题对这类研究所需物质的时间安排和最终数量有重大影响,因此,人们还认为,能够使用灵活尺度的流动化学方法按需生产是非常可取的。图1自1835年费希尔首次合成吲哚类化合物的报告以来,又有十几种独特的吲哚类化合物被报道出来,显示出在这些有价值的结构中开发新化合物的重要性。然而,大多数吲哚类物质合成的共同特点是需要使用有害物质,如肼(Fischer)、重氮类化合物(Japp Klingemann)或叠氮化物(Hemetsberger Knittel),或者在吲哚环形成之前必须在多步反应中构建特定功能的前体。为了克服这些潜在的缺点,Marcus Baumann及其团队开始开发一种良性的工艺,依靠廉价的底物和无毒的试剂,用一种容易扩大且连续的方式迅速合成所需的吲哚。为了通过稳健的合成步骤生成核心吲哚单元,University of Durham化学系的Ian R. Baxendale教授团队决定研究以氰乙酸乙酯(10)为亲核试剂,在碱介导的SNAr反应中处理2-氯硝基苯9(Scheme1)。所得到的加合物11将在非均相氢化条件下,通过还原环化步骤生成吲哚产物12。从相应的酯功能化吲哚12,预期与肼缩合会得到相应的酰肼13,其可通过活性羰基供体的作用如CDI(1, 1-羰基二咪唑)或三光气(双(三氯甲基)碳酸酯)环构化得到所需的产品8。这种合成策略具有多种优势,因为它依赖于现成的底物和试剂,产生少量无毒副产物(碱?HCl、H2O),并在关键的环构化步骤中使用工业上较友好的加氢工艺。相关成果喜人,并于2017年发表在《BeilsteinJournal of Organic Chemistry》杂质上(Beilstein J. Org. Chem. 2017, 13, 2549–2560)。为了开始这项研究,作者首先进行了一个全面的筛选流程,以确定形成化合物11的流动兼容性条件。对溶剂、碱、温度和试剂化学计量比的优化选择结果如表1所示。表1虽然DMF和乙腈是很好的反应溶剂(表1,第3,6,10,15,23和26项),但在淬灭反应(1 M HCl)后有效地提取产品上遇到了困难。乙酸乙酯性能优异,可使萃取非常容易,但在反应过程中观察到少量暗红色且粘稠的沉淀物(表1,第12项)。在中试尺度的流动反应器工艺中这被认为是有问题的,因为可能造成堵塞。然而,研究发现,添加10~20% v/v的乙腈可以保证溶液完全均匀,也可以进行简单的水萃取,回收率较高(>90%)。然而,从初步结果来看,DCM是最有效的溶剂(表1,第7、24和25项),允许0.25 M的底物9的溶液在50℃时用TMG或DBU(2.5当量)和氰乙酸乙酯(10,1.1当量)进行定量转化。基于这些筛选结果,作者设计了一个简单的流动装置,两股预备的原料液在一个简单的T型混合器中汇合,随后直接到一个维持在50℃的加热的流动线圈反应器中(Scheme 2)。迅速形成的深红色溶液(SNAr离子加合物)在保温周期(35 -108 min)后使用在混合后的混合物相分离之前,通过专用的混合芯片进行混合的第三股物料盐酸(1 M)淬灭。为了使相分离成为可能,作者使用了一个基于改进的Biotage通用分离器的被动膜系统。这使得较重的氯化物相可以从较低的连接处去除,较轻的水相可以从溢流位置流出。当从主反应器流出的流量为0.5-1.2 mL/min时,该装置可靠地进行了良好的淬灭和分离。然而,在高流速下产生了两相混合物的不完全分离(有些乳液形成),导致产品中的物质向水中流失从而造成损失,这是由于在较高的流速下在线搅拌芯片产生了较大的剪切力,以及形成高度柱塞流所需要的稳定时间更长。这个问题可以通过将水从第一个分离器引出到第二个等效装置,或者简单地通过两个平行分离器将原来的淬灭流股分开来解决。虽然这些方法在设计或使用分离器组件时不是最优的,但在功能上是可行的。因此,作者还研究了用各种配置的T型和Y型连接器替换有问题的混合芯片,但这立即带来了其他问题,因为不完全淬灭导致了产品回收不良和相关的污染。一种更直接的方法是在分离器之前引入一个流动分层区,这是通过简便地引入一段内径更宽的管(从内径1.6 mm→4.0 mm×20 cm的反应器扩展而来)来实现的。这使得反应器可以通过2.0 mL的流量(对于1.0 mL以上的流量,则向系统中添加第二个52 mL的流动盘管)成功地在线淬灭反应,然后由单个Biotage通用分离器依次处理。在所有情况下,主反应器的出料都显示出定量转化,只有在溶剂蒸发后产品被分离出来。反应器流出速率为1.8 mL/min,这使得在稳态条件下的最大吞吐量达到27 mmol/h。尽管该反应器功能多样,但其生产效率低于理想要求,不幸的是,DCM被证明对以下氢化步骤是一个不兼容的溶剂。虽然溶剂交换是可能的,但作者确定,当乙氧基乙酸作为溶剂并在乙酸作为添加剂存在下用乙醇稀释时,可使吲哚的还原环构化得以成功进行。在此基础上,研究了乙酸乙酯中中间体11的放大反应。在之前成功使用Coflore,AM技术公司ACR设备处理流动泥浆后,作者决定在合成中使用该设备来克服在溶解性方面遇到的问题。起始物料用乙酸乙酯中分别储备的料液来制备,而后混合,首先是碱和氰乙酸乙酯(10)在芳氯9的溶液汇合前混合(注意这是一个放热过程),然后进入ACR,振动频率设置为8 Hz(Scheme 3)。为了加强这一过程,并以减少溶剂量为具体目标,以便在随后的还原步骤(乙醇稀释后)中保持一个可行的反应浓度,作者进行了一系列浓度和流速优化。最终发现,在3 mL的混合流速下,相当于停留时间约为30 min,反应器温度为65℃,能够实现1.5 M底物9溶液的定量转化。连续运行时,反应器的出料为暗红色悬浮液,其中包括体积分数约为10%的固体,但即使在较长时间内(> 14 h)也能很容易地通过系统进行处理。该装置的理论工作效率为0.108 mol/h。接下来,为了便于整体淬灭和后处理,作者在以6 mL/min的流速输送的2 M HCl水溶液的汇合点添加了一个静态混合元件(Scheme 4)。通过将暗红色的反应混合物转化为收集时快速相分离的浅黄色双相溶液,可以立即观察到有效淬灭的迹象。通过对有机相的1H NMR和LC分析,确认了该方法淬灭成功,显示只有产物11和微量氰乙酸乙酯(10)。最后进行人工分离,然后在Na2SO4上干燥,将溶剂蒸发,得到了目标产物11,收率94-96%,基于每隔20 min取一次样,共取6次。几种实现间歇分离自动化的实验室方法已经被报道,利用机器视觉系统和采用光学或感应电导率(阻抗测量)来确定相位划分的内联检测设备来确定相分离。然而,由于特定项目的时间限制,作者被迫使用更人工的方法,但肯定会将这种节省劳动力的设备纳入未来的发展计划。作者的基础系统使用一个简单的间歇式收集容器和两个位置合适的高效液相色谱泵来独立地去除水相和有机相。偶尔的人工干预允许间歇重新校准泵,以保持工作体积,并创建一个合适的相段进行去除。同样,在溶剂蒸发后,目标分子11的分离收率高达93-97%。表2水相淬灭分离后收集的溶液中含有0.4 M产品11,该产品11再用乙醇稀释,以提供0.2 M的原液,用于下一个还原性成环步骤。在全氢条件下,使用含10 mol% Pd/C催化剂的ThalesNano H-cube系统进行还原。流速为0.4 mL/min,温度为50℃,完全转化为吲哚产物12(表2,第1项)。当流速提高到0.8-1 mL/min时,观察到原料完全消耗,但分离出混合产物(表2,第2、3项)。对其组成的详细分析表明了所需产物(12,62%)和另外两种化合物(图2)的存在,这两种化合物在柱层析分离后分别被确认为氨基吲哚(14,29%)和n-羟基氨基吲哚(15,9%)。正如预期,随着流速进一步增加,n-羟基氨基吲哚15的比例增加,后两种不完全还原产物增多。这与以往文献中使用催化氢化或化学计量金属(锌或铟)介导还原的研究结果相关联,并且与逐步还原机制相一致(Scheme 5)。从实验序列中可以明显看出,需要几倍当量的氢才能完全还原,因此较高的氢浓度(压力)是有益的(表2,第4-10项)。作者还注意到,根据机理,苯胺氮的质子化可以促进还原步骤,并有助于水(16和18)和氨(20)的去除。因此,作者筛选了一系列酸性催化剂,其中乙酸(10-30 mol%)最佳(表2,第11-14项)。较强的酸或较高的酸负荷往往会导致产生较高的内部压力,在某些情况下,若观察到沉淀物的形成,则需要将运行提前终止并安全关闭。使用乙酸并限制酸的浓度(10 mol%),在较高的内部压力(15 bar)下运行,能够稳定且连续地操作,流量允许提高到1.3 mL/min,同时确保>98%的转化至所需的吲哚12(表2,第15-18项)。这转化为15.6 mmol/h(3.7 g/h产品)的通量。通过9:1正己烷/乙醚溶剂蒸发和研磨,可分离出白色固体最终产物,收率93%。这种净化除去了残留的乙酸,杂质即使存在也只是痕量的副产物。图2在接下来的实验序列中,作者研究了这个过程的最后两个步骤:乙氧基取代物的联氨,然后成环形成3H-[1, 3, 4]恶二唑-2-1单元。在此过程中,将化合物12的0.95 M THF溶液与肼溶液(THF中,1.0 M)混合,直接进入加热的流动线圈中,在100℃下进行过热(Scheme 6)。停留时间为40 min,可以完全转换为相应的酰肼13,该酰肼13与含有CDI(THF中,1.1 M)的另一股料液直接混合,并在第二个流线圈中75℃加热26 min。这提供了最终产品8的定量转换,产品流通过QP-SA清除盒(一种磺酸功能化聚合物)易于提纯。该产品8经溶剂蒸发得到黄色固体(94%),但需要从DCM重结晶得到白色非晶粉末,纯度高,收率82%。作者进行了一系列在最后一步中使用碳酸二甲酯作为CDI替代试剂的尝试,在一系列条件下都失败了,而在前面的酰肼形成步骤中直接使用(甲氧羰基)肼(CAS:6294-89-9)的试验也失败了。然而,作者发现三光气(0.4当量)可以成功地应用于后者的成环过程。使用与Scheme 6类似的反应器组件,将三光气(氯仿中,0.8 M)与中间体13的溶液混合,通过一个加热(55℃)的流动线圈,然后通过一个含有QP-DMA(N, N-二甲基苄胺聚苯乙烯)的填充床扫气筒。溶剂蒸发后,产品为白色固体,分离收率91%。在实践中,这一方法被证明远远优于以前使用的CDI环化直接从反应器获得更高的总收率和质量提升的产品。实际上,最终产品8的纯度足够直接应用,允许在反应装置中用总结:1.按照文中所述的步骤,作者已经能够通过一个灵活的实验流程快速制备所需的目标分子8;2. 在整个实验流程中,英国AM Technology公司的连续多级搅拌反应器Coflore ACR在第1步生成中间体11的反应中起到了关键作用,因为反应中会不断生成大量不溶的固体盐副产物,而Coflore ACR可以在防止堵塞的同时使反应连续稳定运行;3. Coflore ACR反应器可以解决连续流反应中的多项问题,包括对反应的高效淬灭以及处理含不溶性固体的反应,体现了Coflore ACR反应器良好的实用性能;4. 作者利用Coflore ACR反应器实现了第1步中间体11的连续化稳定生产,并与整个后续流程实现串联,表明不溶性固体不再是限制连续流生产的问题,该项技术可以在精细化工产品的大量制备中起到关键性作用;5. Coflore ACR是已经成熟商业化的反应器,具备很好的规范性和通用性,对如含固等多相等复杂体系的工业化应用前景十分广阔。参考文献:doi:10.3762/bjoc.13.251Beilstein J. Org. Chem. 2017, 13, 2549–2560.公司简介:深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司(连续多级搅拌反应器、催化加氢系统)、英国NiTech公司(连续结晶仪、连续合成仪)在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
厂商
2019.07.01
液滴加速反应筛选法指导地西泮的多步连续流合成及对反应机理的快速质谱分析研究
众所周知,质谱(MS)对于反应监测方面有着大的用途,其在监测反应动力学以及反应结果方面具有优良的特性,并且MS也有助于识别反应中间体,从而了解化学反应的机理细节。此外,质谱还可以用来研究液滴中的化学反应。以往的研究表明,在电喷雾电离(ESI)形成的微滴中,大多数化学反应的反应都比相应的本体反应速率加快。这种加速现象产生的一部分原因是溶剂蒸发及其对界面处试剂浓度的所带来的影响。这种快速反应的现象可作为筛选方法用于指导微流体与大量反应。利用微滴研究化学反应的另一种方法是利用莱登弗罗斯特效应(Leidenfrost effect)。其原理是当液滴接近被加热的表面时,溶剂开始蒸发,在液滴周围形成一层隔绝蒸汽层。这层防止了溶剂的快速蒸发,并导致液滴悬浮。尽管通过该技术形成的液滴比ESI液滴大,但它们具有一些相同的性质;因此,该液滴中的反应也可以成为指导微流体反应的有效工具。Purdue大学的H. Thompson教授2017年通过使用地西泮的合成(图1)作为模型系统来说明液滴筛选如何指导地西泮的连续合成,相关成果发表在Org. Process Res. Dev.期刊上。由于其途径中的一个关键步骤涉及N-酰化,因此该成果也可能可以用于指导合成普通生物活性分子中的N-酰化反应,因此具有更深远的意义。虽然微滴中的反应条件并不总是可以直接等同于为微流体级的反应条件,但该方法仍可以作为一种快速的判断预测工具,以预测该条件下,反应在生产级微流体体系中发生的可能性。流动化学系统,再加上ESI-MS的在线监测,能够通过实时反馈快速筛选反应条件。这不仅使原料药的合成更加简便、高效,而且也为人们对反应机理和副产物形成的新认识提供了可能。地西泮可以通过两步合成获得,从5-氯-2-(甲胺基)二苯甲酮(1)与2-卤乙酰氯(2)的N-酰化反应开始,得到酰胺(3)。随后用氨水处理,然后进行环化,得到地西泮4(图1)。对于在液滴中筛选或在流动反应系统中检查每个反应,通过ESI-MS分析立即确定结果。图1 地西泮的两步合成N-酰化反应筛选。反应筛选从ESI液滴、莱登弗罗斯特液滴、大量反应和流动反应系统中的N-酰化步骤开始。最初的ESI喷雾(脱机)反应在多个溶剂[二甲基乙酰胺(DMA),四氢呋喃(THF),二甲基甲酰胺(DMF),乙腈(ACN),N-甲基吡咯烷酮(NMP),甲苯]中进行筛选,结果显示相对于相同起始浓度进行的大量反应进行30分钟。在ACN(35×)和甲苯(100×)中反应加速很明显。虽然电喷雾实验是在不同的溶剂中进行的,但在所有实验中分析之前的最后提取操作都是在ACN中进行的。图2 在甲苯和乙腈(ACN)体系中对于N-乙酰化反应的MS分析第一步的流动反应条件筛选也从溶剂筛选开始。NMP和DMF表现不佳,转化率有限,杂质较多。在甲苯中,氯化反应发生得很快,转化率很好,而在ACN中,观察到由SN2反应途径产生的副产物(5,7)的形成(图2)。除了溶剂,也对温度和反应时间进行了筛选。随着温度的升高,在大多数情况下,由于副反应越来越多,转化率变底。通过将氯乙酰氯更换为溴乙酰氯,作者找到了副反应发生的机理证据。使用溴乙酰溴的N-酰化反应的预期产物的分子量为367;但是,在m/z 322处看到了一个额外的峰。这证实了在N-酰化过程中溴而非氯的损失。通过柱色谱分离出SN2和N-酰化产物的混合物(来自流动实验),核磁共振分析显示,在3.7和3.9 ppm处有两组不同的峰,分别对应于N-乙酰化和SN2产物的亚甲基质子(图3)。根据上述观察,我们确定甲苯是连续合成地西泮的最佳溶剂。图3 SN2反应和N-乙酰化反应产物混合物的亚甲基质子的1H NMR信号除此SN2产物外,在流动实验中还观察到m/z 288处的峰,与环闭合的产物一致。作者认为,这种m/z 288化合物(分子量287)是是由于SN2反应后,羰基对酰氯的亲核攻击形成七元环状内酯的产物。实验结果表明,在微流体系统中,反应溶剂和卤化物的选择可以对反应结果起到控制作用。值得注意的是,这些副反应的结果在微流体实验中观察到,但在相应的液滴实验中却没有。这是由于微流控反应中需要高温和高压条件,但液滴反应却不需要,原因可能是副反应需要更高的能量。环化反应筛选:对环化反应的预筛选显示,在莱登弗罗斯特液滴中形成地西泮需要更高浓度的氨(图4)。在这些液滴反应中可以观察到反应的加速。在尝试两步连续流地西泮合成之前,首先对先前制备的N-酰化产物3A的第二步进行了微流体筛选。对每种溶剂,筛选其不同的反应温度和反应时间。该物质在ACN和甲苯溶解度小于250μM,但在该浓度下在NMP中溶解良好。幸运的是,在NMP中地西泮的转化效果很好。在研究这一步骤时,另一个有趣的现象是在m/z 303处出现质谱峰。这可能代表甲基位置发生了氮的取代。由于在较低的温度和反应时间中MS中出现这一峰,在较高的温度下消失,所以说这可能是地西泮合成的中间产物。LC-MS分析表明,在整个温度和反应时间筛选中实验中,即使地西泮的数量稳步增加,这种m/z 303的物质的数量也依旧保持相对恒定,这有可能是由于m/z 303的物质的高电离效率淹没了反应混合物中其他化合物的信号。该m/z 303的物质(6)也被认为是地西泮合成中的水解产物。图4 环化反应筛选中在ACN和甲苯溶剂中使用溴乙酰氯对地西泮合成的结果比较连续流地西泮合成:作者通过根据上述的实验结果,进而对地西泮合成路线中的每一个合成步骤有了更全面的了解。这些可能的反应途径总结在图5中。在筛选的指导下,作者接下来尝试通过两个连续步骤优化地西泮的合成。通过使用了一个Two-chip反应器系统来更好地控制每个chip的温度和反应时间。第一chip分别以1:2的比例(图6、R1和R2)将5-氯-2-(甲胺基)二苯甲酮和卤代乙酰氯组合在一起,然后将所得N-酰化产物混合物(R3)与甲醇稀释并随后在第二chip中以7倍的过量添加氨/甲醇(R4)。图5 在地西泮合成中可能存在的一些副反应途径溶剂筛选再次受到溶解度的限制,尤其是在第二步中添加氨/甲醇时。为了缓解这一问题,在第一反应步骤后加入稀释步骤以提高溶解度。第一步使用甲苯,用甲醇或NMP稀释1:4,可获得良好的溶解性。第一步使用ACN和稀释也是有效的。与之前的流动反应的筛选一样,作者改变了温度和反应时间,以及溴乙酰氯和氯乙酰氯的选择。这次筛选的结果证实了作者以前的观察,特别是在ACN中使用溴乙酰氯容易产生了一些SN2产物。此外,使用溴乙酰氯时,从中间体到地西泮的总转化率较高。图6采用Chemtrix Labtrix S1全自动微通道连续流反应器两步连续流合成地西泮的装置系统示意图每个反应的产率使用定量ESI-MS/MS方法进行测定。采用ESI-MS定量方法,在甲苯/甲醇溶剂体系中使用溴乙酰氯,最终以100%产率获得地西泮。这也是Leidenfrost和喷雾微滴中的最佳溶剂体系。实验结论1.本研究以地西泮合成为模型反应,展示了MS分析和液滴反应对微流体合成的指导作用。MS不仅可以作为一种分析工具,而且可以作为一种快速预测反应和指导微观合成的方法。2.使用喷雾和莱登弗罗斯特液滴反应作为筛选中的一步,以指导更大规模的微流体筛选的策略,用于预测反应的总体结果是有效的。虽然在液滴实验的结果与流动反应中所观察到的有一些细微差别,但这些实验仍然可以提供给我们反应是否可行的信息。3.通过在微流控反应器中分两步连续合成地西泮。其特点是使用混合溶剂系统,以及按顺序排列的两个微流控芯片,从而在每个步骤实现对温度控制的优化。4.文中涉及微反应采用荷兰Chemtrix 公司的Labtrix S1 自动微反应系统中3223和3224配置执行。5.作者还发现了地西泮合成中在之前未知的副反应途径。这些结果展示了微流体合成与快速ESI-MS分析相结合的可能性,以识别先前未知的反应途径并指导优化原料药的连续合成。参考文献Org. Process Res. Dev. 2017, 21, 1566?1570公司简介:深圳市一正科技有限公司,作为荷兰Chemtrix公司(微通道反应器)、英国AM公司(连续搅拌多级反应器、催化加氢系统)、英国NiTech公司(连续结晶仪、连续合成仪)在中国区的独家代理商和技术服务商,为广大高校和企业提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、南京大学、中山大学、华东理工大学、南京工业大学、浙江工业大学、河北工业大学等高校研究机构合作成立微通道连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。公司与荷兰Chemtrix B.V.在浙江台州、江苏南京合作组建了连续流微通道工业化应用技术中心(以下简称“工业化技术中心”),旨在打造集连续流微通道工艺开发、中试试验、工业化验证、技术交流于一体的综合性连续流微通道应用技术服务中心,以为广大生物医药企业、化工类企业提供专业、完善的智能化连续流工艺整套系统解决方案及一流的技术服务方案。
厂商
2019.06.04

连续流化学技术交流会
连续流化学技术交流会邀请函 近些年,连续流反应技术已经从小众的学术应用研究转化为一种公认的工业技术。其优势在于该技术可以提高对热效应的处理能力、增强混合能力和更大的操作窗,从而能够开发出更安全、高效、稳健和可持续的合成生产工艺,并且在生产中所带来的效益不仅限于提高工艺安全性,还包括实现更高效、更低成本的工艺,从而降低下游加工成本和人力成本。其次实验室和生产规模上的应用意味着连续流反应器技术能够得使得早期研发人员与生产工程师双双受益,促进安全、高效和可持续工艺的发展,这将对化学工业的发展产生重大影响。 一、会议主办单位深圳市一正科技有限公司、荷兰Chemtrix B.V 公司二、会议主题连续流化学技术交流会三、会议费用免费四、会议内容 报告时间报告题目报告人13:30-14:15连续流化学-从研发概念到工业化规模生产实施Dr. Charlotte WilesChemtrix BV 14:15-15:00医药合成新技术:流动化学邓建 博士清华大学化工系15:00-15:45气液固三相的连续流工艺整体解决方案钟明深圳市一正科技有限公司15:45-16:30精细化学品的连续化工艺研究与创新万力 博士华东理工大学 五、会议时间2019年 6月18日下午13:30-16:30六、会议地点展馆地点:上海新国际博览中心(上海市浦东新区龙阳路 2345 号,地铁 7 号线花木路站)技术交流会地点:N3M43会议室(近N3馆)展台位置:N1A15(1号馆2号大厅入口第一排) 展出产品: 七、报告人简介报告人:Dr. Charlotte Wiles Dr. Charlotte Wiles, Chemtrix BV 公司首席执行官,2003 年获得英国 Hull 大学博士学位, 博士期间研究微反应器在有机合成化学中的应用,在微反应器合成领域中有超过 10 年的研究经验。 期间以微反应器为主题发表了大量的 SCI 文章、评论及著作并成功申请多项专利。现为英国皇家化学协会会员,美国化学协会会员,美国化学工程师协会会员。报告人:邓建2003年获得兰州大学化学化工学院化学学士学位,2008年获得兰州大学化学化工学院高分子化学与物理博士学位(与北京大学化学与分子工程学院联合培养)。2008年11月至2010年9月,在美国威斯康辛大学密尔沃基分校从事博士后研究工作。回国后,2011年至2017年六年期间,一直在医药企业(暨明医药,爱斯特生物制药和乐威医药)担任技术研发主管,主要负责客户沟通、团队管理、原料药和中间体的工艺开发、过程管控和车间生产。2017年,以科研助理加入清华大学化工系,从事流动化学技术的工业应用研究。2019年,正式被清华大学聘为助理研究员,研究方向为流动化学技术与工程。具体研究内容包括:微型/小型反应器中化学反应新规律,连续绿色安全合成精细化学品(原料药、农药、含氮含能材料和功能聚合物材料),流动化学技术工业应用,流动化学工艺研发和工业应用方法学。迄今为止,发表SCI收录论文7篇,英文综述1篇,已公开和授权专利4项。在企业工作期间,被评为四川省“千人计划特聘专家”,负责的“5-氟胞嘧啶合成新工艺研发”项目获得2013年成都市科技进步三等奖。曾任长虹虹视、成都有机所和成都爱斯特成立的有机联合发光材料实验室执行主任,联合日韩OLED材料终端用户,开发新型OLED发光材料。报告人:钟明深圳市一正科技有限公司副总经理,一正科技是荷兰Chemtrix公司(微通道反应器)、英国 AM公司(连续搅拌多级反应器、催化加氢系统)、英国AWL(连续过滤干燥仪)在中国区的独家代理商和技术服务商,提供连续合成、在线萃取、连续结晶、在线过滤干燥、在线分析等整套连续工艺解决方案。公司与复旦大学、中山大学、南京大学、华东理工大学、浙江工业大学、河北工业大学等高校研究机构合作成立连续流化学联合实验室,致力于推动连续流工艺在有机合成、精细化工、制药行业、能源材料、食品饮料等领域的应用,合作实验室可以为客户的传统间歇釜式工艺在连续流工艺上的转变提供工艺验证、连续流工艺开 发工作,促进制药及精细化工企业由传统间歇工艺向绿色、安全、快速、经济的连续工艺转变。报告人:万力万力博士主要从事高性能化学品的快速精准绿色化制造,以微通道技术为基础,结合微波与超声波等方式创新化学品合成工艺,并实现萃取、分离、过滤等后处理连续化操作。同时基于合成工艺特征,设计和制造匹配型微化工装置,构建智能化系统化微化工生产平台,精准控制进料、反应监测、后处理等环节,有效降低生产成本和废弃物排放,提升生产安全性。目前作为项目负责人已承担4项国家和省部级自然科学基金,承担多项企业合作项目。在J.Am.Chem.Soc.,Angew.Chem.Int.Ed.,Adv.Syn.Cat.,Green. Chem.等期刊发表SCI论文16余篇,授权中国发明专利4项。八、 联系方式联系人:袁女士联系方式:0755-83549661/15915410392电子邮箱:info@e-zheng.com 九、 参会人登记信息(扫描下方二维码进行预约登记) 备注:同单位参加人数不超过2人。 深圳市一正科技有限公司 2019年5月21日
厂商
2019.05.30
公司名称: 深圳市一正科技有限公司
公司地址: 深圳市宝安区飞扬科技园A栋510 联系人: 李小姐 邮编: 518000 联系电话: 400-860-5168转1639
仪器信息网APP

展位手机站