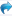所有提问
色谱
|
光谱
|
质谱
|
波谱
|
显微镜
|
物性测试
|
样品前处理
|
常用设备
|
食品检测
|
药物分析
|
环境监测
|
实验室建设/管理
|
认证认可
|
基础知识
更多>>
未解决的问题:
146802
所有仪器问答:
170764
您现在的位置:
首页
>
仪器问答
>
色谱
>
液相色谱
液相色谱论坛
共有 23 人回复了该问答
求助液相色谱进样品不出峰原因分析
回复
shixiangqun
发表于:2010/10/27 21:12:48
悬赏金额:
100积分
状态:
未解决
一、仪器:Varian HPLC 230泵/325紫外检测器(单波长)、配国产柱温箱、手动进样、
Varian C
18
(
5μm
,
4.6mm×150mm
).
二、实验过程:测丹参片中丹参酮IIA溶出度
1、配系列丹参酮IIA标准溶液(溶剂用85%甲醇),共5个标样,先在紫外分光光度计上进行光谱扫描,峰值检出,在269.3nm有最大吸收峰,再做定量测定,标准曲线相关系数为0.998,溶出介质为稀盐酸,溶出样品按时间点分别取样7个,针筒过滤器过滤,不稀释上紫外测试,30分钟前溶出不明显,测出吸光度值低,30分钟后有明显溶出,吸光度值高,位于标3与标4之间。
下面是标样的及溶出样品的紫外定量测定图:(见17楼的补充贴)
2、溶出条件不变,溶出样品采用液相进行测试,液相条件:检测波长270nm;流动相:85%甲醇:15%水;等度洗脱,分析时间10分钟;柱温:30度;流速:1ml/min。
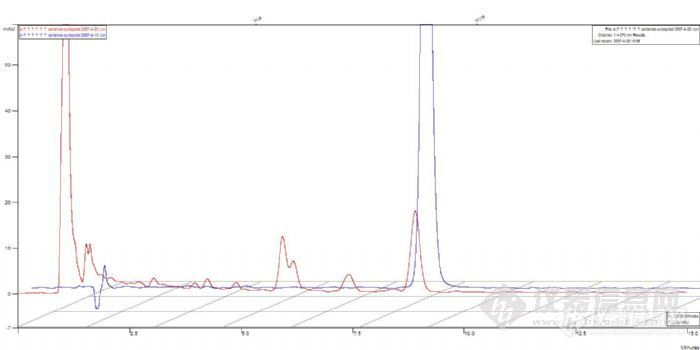
3、取丹参酮IIA标准溶液(溶剂用85%甲醇),共5个标样,分别依次手动进样,进样量100uL(定量环20uL),标样均在5.4min左右出峰,各标样保留时间非常重合,对照品标准曲线相关系数:0.996。
下面是5个标样的色谱图,色谱图进行了偏置2%,为了利于版友观察,从前往后分别是标1-5:
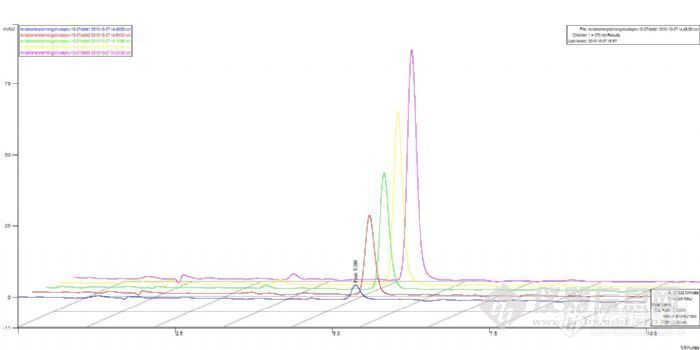
下面的图是标准曲线图,标1的峰面积有的偏小,但还是基本成线性:
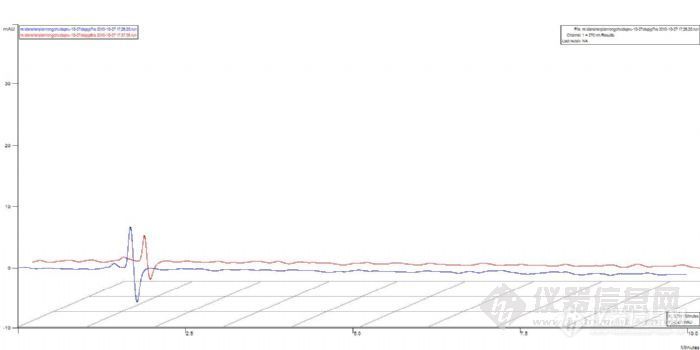
4、前面实验过程都比较顺利,在预想在范围内,问题出现在最后时刻,开始对溶出的7个时间点样品(直接从溶出杯中取样,溶剂为稀盐酸,针筒过滤器过滤)上HPLC进行测定,怪事发生了,竟然没出有一个出峰的,连一个其它的杂峰都没有,只有开始时的一个溶剂峰。
下面是7个溶出样品的色谱图,色谱图进行了偏置2%,为了利于版友观察:
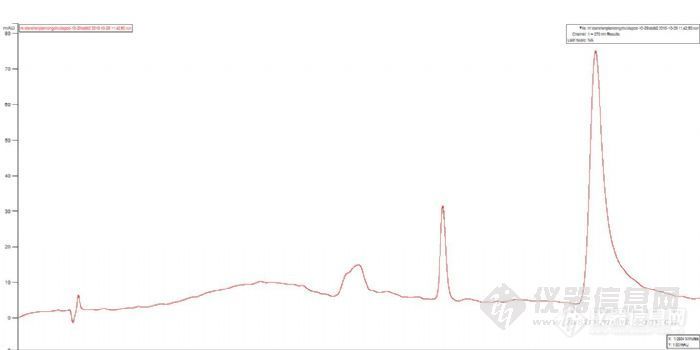
5、原来我们做过丹参片中丹参酮IIA含量的液相测定,方法基本一致,标准曲线比较容易做,样品用85%甲醇超声定容,除了和标样在同样位置出峰外,在这峰之前还有其它成分的峰。
下面是以前做的丹参片样品及丹参酮IIA标样的色谱图,色谱图进行了偏置2%,为了利于版友观察,前面的丹参片,后面的丹参酮IIA:
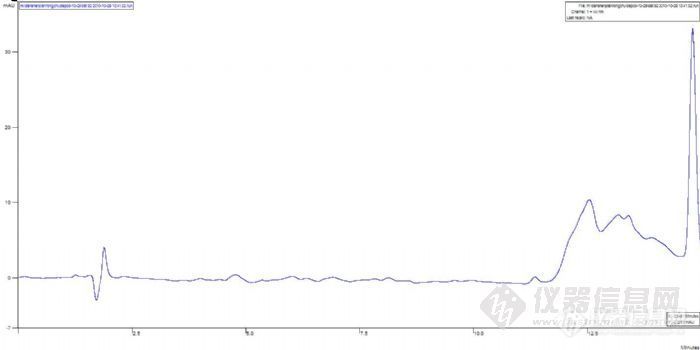
6、我怀疑稀盐酸在其中做怪,于是调整样品配置方法,将第6、7时间点采集的溶出样品各取1mL,用85%甲醇定量到10mL,以图消除稀盐酸的影响,重新配制的第6、7的溶出样品分别进样,结果还是让人晕倒,竟然还是一条直线,除了溶剂峰外,一个峰都没有,跟进一针试剂空白一样,由于今天太晚了,没有再做其它方案调整,于是冲柱2h后走人了,谱图也忘记拷贝了。
下面是用甲醇稀释过的溶出样品6、7的色谱图,色谱图进行了偏置2%,为了利于版友观察:
三、回家后,想想今天真是郁闷啊!本想今天实验十拿九稳,大不了线性不好,但竟然不出峰,让我摸不着头脑,做液相以来,这次让我最纠结,急盼高手指点迷津,万分感谢!
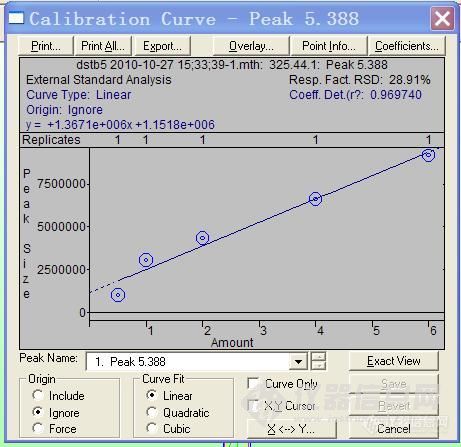
四、第二天调整了色谱条件,同等度洗脱改为梯度洗脱,甲醇由60%梯度到90%,先进了一针标样,看出峰靠后,于是增加了分析时间,又进了一针同样的标样,但结果都让人不满意,标样出峰很怪,标样峰前面的驼峰正常吗?
下面是梯度洗脱方法做的两次标样,两次的分析时间不同,第一针是15分钟,第二针改为了20分钟:
五、再次调整了色谱条件,改变梯度洗脱方式,先前甲醇由60%梯度到90%,改为甲醇由70%梯度到95%,进了一针溶出样品,看是否有出峰?
下面是改变梯度洗脱方法进的一针溶出样品,分析时间是20分钟:
六、问题不但没有解决,反而梯度洗脱又出了让人看不懂的色谱图,第一次梯度的色谱图中的驼峰还可以理解成梯度洗脱使得基线上漂,而第二次梯度的色谱图中驼峰与目标峰中间竟然又多出了一个峰,无法理解,再次陷入了困局,望高手再度出山,来拨云见日,多谢了!
主贴内容在不断更新,望各位版友能持续观注!!!
·
·
·
·
共有 23 人回复了该问答
求助液相色谱进样品不出峰原因分析
回复
shixiangqun
发表于:2010/10/27 21:12:48
悬赏金额:
100积分
状态:
未解决
一、仪器:Varian HPLC 230泵/325紫外检测器(单波长)、配国产柱温箱、手动进样、
Varian C
18
(
5μm
,
4.6mm×150mm
).
二、实验过程:测丹参片中丹参酮IIA溶出度
1、配系列丹参酮IIA标准溶液(溶剂用85%甲醇),共5个标样,先在紫外分光光度计上进行光谱扫描,峰值检出,在269.3nm有最大吸收峰,再做定量测定,标准曲线相关系数为0.998,溶出介质为稀盐酸,溶出样品按时间点分别取样7个,针筒过滤器过滤,不稀释上紫外测试,30分钟前溶出不明显,测出吸光度值低,30分钟后有明显溶出,吸光度值高,位于标3与标4之间。
下面是标样的及溶出样品的紫外定量测定图:(见17楼的补充贴)
2、溶出条件不变,溶出样品采用液相进行测试,液相条件:检测波长270nm;流动相:85%甲醇:15%水;等度洗脱,分析时间10分钟;柱温:30度;流速:1ml/min。
3、取丹参酮IIA标准溶液(溶剂用85%甲醇),共5个标样,分别依次手动进样,进样量100uL(定量环20uL),标样均在5.4min左右出峰,各标样保留时间非常重合,对照品标准曲线相关系数:0.996。
下面是5个标样的色谱图,色谱图进行了偏置2%,为了利于版友观察,从前往后分别是标1-5:
下面的图是标准曲线图,标1的峰面积有的偏小,但还是基本成线性:
4、前面实验过程都比较顺利,在预想在范围内,问题出现在最后时刻,开始对溶出的7个时间点样品(直接从溶出杯中取样,溶剂为稀盐酸,针筒过滤器过滤)上HPLC进行测定,怪事发生了,竟然没出有一个出峰的,连一个其它的杂峰都没有,只有开始时的一个溶剂峰。
下面是7个溶出样品的色谱图,色谱图进行了偏置2%,为了利于版友观察:
5、原来我们做过丹参片中丹参酮IIA含量的液相测定,方法基本一致,标准曲线比较容易做,样品用85%甲醇超声定容,除了和标样在同样位置出峰外,在这峰之前还有其它成分的峰。
下面是以前做的丹参片样品及丹参酮IIA标样的色谱图,色谱图进行了偏置2%,为了利于版友观察,前面的丹参片,后面的丹参酮IIA:
6、我怀疑稀盐酸在其中做怪,于是调整样品配置方法,将第6、7时间点采集的溶出样品各取1mL,用85%甲醇定量到10mL,以图消除稀盐酸的影响,重新配制的第6、7的溶出样品分别进样,结果还是让人晕倒,竟然还是一条直线,除了溶剂峰外,一个峰都没有,跟进一针试剂空白一样,由于今天太晚了,没有再做其它方案调整,于是冲柱2h后走人了,谱图也忘记拷贝了。
下面是用甲醇稀释过的溶出样品6、7的色谱图,色谱图进行了偏置2%,为了利于版友观察:
三、回家后,想想今天真是郁闷啊!本想今天实验十拿九稳,大不了线性不好,但竟然不出峰,让我摸不着头脑,做液相以来,这次让我最纠结,急盼高手指点迷津,万分感谢!
四、第二天调整了色谱条件,同等度洗脱改为梯度洗脱,甲醇由60%梯度到90%,先进了一针标样,看出峰靠后,于是增加了分析时间,又进了一针同样的标样,但结果都让人不满意,标样出峰很怪,标样峰前面的驼峰正常吗?
下面是梯度洗脱方法做的两次标样,两次的分析时间不同,第一针是15分钟,第二针改为了20分钟:
五、再次调整了色谱条件,改变梯度洗脱方式,先前甲醇由60%梯度到90%,改为甲醇由70%梯度到95%,进了一针溶出样品,看是否有出峰?
下面是改变梯度洗脱方法进的一针溶出样品,分析时间是20分钟:
六、问题不但没有解决,反而梯度洗脱又出了让人看不懂的色谱图,第一次梯度的色谱图中的驼峰还可以理解成梯度洗脱使得基线上漂,而第二次梯度的色谱图中驼峰与目标峰中间竟然又多出了一个峰,无法理解,再次陷入了困局,望高手再度出山,来拨云见日,多谢了!
主贴内容在不断更新,望各位版友能持续观注!!!
·
·
·
·
回复
1#
xue2009
回复于:2010/10/27 23:28:34
是不是样品出现什么问题?
扫一扫查看全部
23
条回复
高级回复
快速回复
【花三五分钟,帮别人解决一个问题,快乐自己一天!】
未解决问题
液相色谱求助
岛津14C想加个六通阀
HPLC测定heme和原卟啉I
维生素B6峰有峰前沿
RP—amide柱压力高
关于液相流动相的细节
柱子堵了怎么解决?
液相色谱泵压为什么一直
cds初始积分事件可以
急问紫外吸收问题
相关仪器
相关资料



 您现在的位置:
您现在的位置: